anvi-script-fix-homopolymer-indels
Table of Contents
Corrects homopolymer-region associated INDELs in a given genome based on a reference genome. The most effective use of this script is when the input genome is a genome reconstructed by minION long reads, and the reference genome is one that is of high-quality. Essentially, this script will BLAST the genome you wish to correct against the reference genome you provide, identify INDELs in the BLAST results that are exclusively associated with homopolymer regions, and will take the reference genome as a guide to correct the input sequences, and report a new FASTA file. You can use the output FASTA file that is fixed as the input FASTA file over and over again to see if you can eliminate all homopolymer-associated INDELs.
🔙 To the main page of anvi’o programs and artifacts.
Authors

Can consume
Can provide
Usage
This program takes an input fasta file with one or more sequences, then corrects INDELs associated with homopolymer regions given a reference fasta file, and reports edited sequences as a new fasta file.
You must be extremely careful with this program since it reports edited sequences.
Better alternatives
If you need a comprehensive solution to correct your long-read sequencing data for serious applications, you should not use this script, but resort to better alternatives designed to correct frameshift errors.
For instance, Arumugam et al’s solution leverages NCBI’s nr protein database to correct frameshift errors through modified-DIAMOND alignments. Another solution, homopolish by Yao-Ting Huang et al, uses mash sketches to correct minION sequences. You may also want to check proovframe by Thomas Hackl, which aims to correct frame-shift errors in long-read sequencing data.
Motivation
We developed this tool largely to test the impact of INDEL errors associated with homopolymers Oxford Nanopore Technology yields. When there is a high-quality reference genome, this program can align a set of input sequences to the reference, and when it sees something like this in the alignment:
Input sequence ......: ... CGAAAAACG ...
Reference sequence ..: ... CGAAA--CG ...
It can correct the input sequence to look like this, since this would indicate that the additional A nucleotides after AAA were likely due to errors from the long-read sequencing:
Input sequence ......: ... CGAAACG ...
Similarly, if the program sees a case like this:
Input sequence ......: ... CGAAA--CG ...
Reference sequence ..: ... CGAAAAACG ...
The program would correct the input sequence this way, assuming that the lacking A nucleotides were likely due to errors from long-read sequencing:
Input sequence ......: ... CGAAAAACG ...
Please note that INDEL errors associated with homopolymers are only a subset of errors that will casue frameshifts and impact amino acid sequences.
Homopolymer length
The parameter --min-homopolymer-length helps the program to determine what to call a homopolymer region. Please read the help menu for this parameter carefully since it is not exactly intuitive.
The value 3 would be a stringent setting. But you may want to lower it to 2 if you promise to evaluate your output carefully.
The script includes some pre-aligned test sequences for you to see how --min-homopolymer-length influences things. For instance, here is the output for --min-homopolymer-length 2:
anvi-script-fix-homopolymer-indels --test-run \
--min-homopolymer-length 2
* >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Query sequence ...............................: ATCGATCGATCGAAAATCGATCGATCG
Reference sequence ...........................: ATCGATCGATCG-AAATCGATCGATCG
GAPS BEFORE REPEATS OF "A"
===============================================
Query ........................................: AAA
Reference ....................................: -AA
Resolution ...................................: {'action': 'DEL', 'positions': [12], 'nt': 'A'}
Edited query sequence ........................: ATCGATCGATCGAAATCGATCGATCG
Query sequence edited correctly? .............: Yes
* >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Query sequence ...............................: ATCGATCGATCGAAAATCGATCGATCG
Reference sequence ...........................: ATCGATCGATCGAA-TCGATCGATCG
GAPS AFTER REPEATS OF "A"
===============================================
Query ........................................: AAA
Reference ....................................: AA-
Resolution ...................................: {'action': 'DEL', 'positions': [14], 'nt': 'A'}
Edited query sequence ........................: ATCGATCGATCGAAATCGATCGATCG
Query sequence edited correctly? .............: Yes
* >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Query sequence ...............................: ATCGATCGATCGAAA-TCGATCGATCG
Reference sequence ...........................: ATCGATCGATCGAAAATCGATCGATCG
GAPS AFTER REPEATS OF "A"
===============================================
Query ........................................: AA-
Reference ....................................: AAA
Resolution ...................................: {'action': 'INS', 'positions': [15], 'nt': 'A'}
Edited query sequence ........................: ATCGATCGATCGAAAATCGATCGATCG
Query sequence edited correctly? .............: Yes
* >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Query sequence ...............................: A-C-AT-GATCG-AAATCGATCGATCG
Reference sequence ...........................: ATCGATCGATCGAAAATCGATCGATCG
GAPS BEFORE REPEATS OF "A"
===============================================
Query ........................................: -AA
Reference ....................................: AAA
Resolution ...................................: {'action': 'INS', 'positions': [9], 'nt': 'A'}
Edited query sequence ........................: ACATGATCGAAAATCGATCGATCG
Query sequence edited correctly? .............: Yes
* >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Query sequence ...............................: ATCGATCGATCGAAATCGATCGATCG
Reference sequence ...........................: ATCGATCGATCGAA-TCGATCGATCG
GAPS AFTER REPEATS OF "A"
===============================================
Query ........................................: AAA
Reference ....................................: AA-
Resolution ...................................: {'action': 'DEL', 'positions': [14], 'nt': 'A'}
Edited query sequence ........................: ATCGATCGATCGAATCGATCGATCG
Query sequence edited correctly? .............: Yes
The output for the same input sequences with --min-homopolymer-length 3:
anvi-script-fix-homopolymer-indels --test-run \
--min-homopolymer-length 3
* >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Query sequence ...............................: ATCGATCGATCGAAAATCGATCGATCG
Reference sequence ...........................: ATCGATCGATCG-AAATCGATCGATCG
GAPS BEFORE REPEATS OF "A"
===============================================
Query ........................................: AAAA
Reference ....................................: -AAA
Resolution ...................................: {'action': 'DEL', 'positions': [12], 'nt': 'A'}
Edited query sequence ........................: ATCGATCGATCGAAATCGATCGATCG
Query sequence edited correctly? .............: Yes
* >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Query sequence ...............................: ATCGATCGATCGAAAATCGATCGATCG
Reference sequence ...........................: ATCGATCGATCGAA-TCGATCGATCG
Edited query sequence ........................: ATCGATCGATCGAAAATCGATCGATCG
Query sequence edited correctly? .............: No
* >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Query sequence ...............................: ATCGATCGATCGAAA-TCGATCGATCG
Reference sequence ...........................: ATCGATCGATCGAAAATCGATCGATCG
GAPS AFTER REPEATS OF "A"
===============================================
Query ........................................: AAA-
Reference ....................................: AAAA
Resolution ...................................: {'action': 'INS', 'positions': [15], 'nt': 'A'}
Edited query sequence ........................: ATCGATCGATCGAAAATCGATCGATCG
Query sequence edited correctly? .............: Yes
* >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Query sequence ...............................: A-C-AT-GATCG-AAATCGATCGATCG
Reference sequence ...........................: ATCGATCGATCGAAAATCGATCGATCG
GAPS BEFORE REPEATS OF "A"
===============================================
Query ........................................: -AAA
Reference ....................................: AAAA
Resolution ...................................: {'action': 'INS', 'positions': [9], 'nt': 'A'}
Edited query sequence ........................: ACATGATCGAAAATCGATCGATCG
Query sequence edited correctly? .............: Yes
* >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Query sequence ...............................: ATCGATCGATCGAAATCGATCGATCG
Reference sequence ...........................: ATCGATCGATCGAA-TCGATCGATCG
Edited query sequence ........................: ATCGATCGATCGAAATCGATCGATCG
Query sequence edited correctly? .............: No
The output for the same input sequences with --min-homopolymer-length 4:
anvi-script-fix-homopolymer-indels --test-run \
--min-homopolymer-length 4
* >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Query sequence ...............................: ATCGATCGATCGAAAATCGATCGATCG
Reference sequence ...........................: ATCGATCGATCG-AAATCGATCGATCG
Edited query sequence ........................: ATCGATCGATCGAAAATCGATCGATCG
Query sequence edited correctly? .............: No
* >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Query sequence ...............................: ATCGATCGATCGAAAATCGATCGATCG
Reference sequence ...........................: ATCGATCGATCGAA-TCGATCGATCG
Edited query sequence ........................: ATCGATCGATCGAAAATCGATCGATCG
Query sequence edited correctly? .............: No
* >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Query sequence ...............................: ATCGATCGATCGAAA-TCGATCGATCG
Reference sequence ...........................: ATCGATCGATCGAAAATCGATCGATCG
Edited query sequence ........................: ATCGATCGATCGAAATCGATCGATCG
Query sequence edited correctly? .............: No
* >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Query sequence ...............................: A-C-AT-GATCG-AAATCGATCGATCG
Reference sequence ...........................: ATCGATCGATCGAAAATCGATCGATCG
Edited query sequence ........................: ACATGATCGAAATCGATCGATCG
Query sequence edited correctly? .............: No
* >>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
Query sequence ...............................: ATCGATCGATCGAAATCGATCGATCG
Reference sequence ...........................: ATCGATCGATCGAA-TCGATCGATCG
Edited query sequence ........................: ATCGATCGATCGAAATCGATCGATCG
Query sequence edited correctly? .............: No
Tips and Warnings
As you correct your input sequences one round, the BLAST may produce new homopolymers. So you may want to re-run the tool by turning the output sequence into an input sequence. For instance, we had a genome reconstructed using long-read sequencing that matched to a gold-standard genome on NCBI.
Running the script the first time this way,
anvi-script-fix-homopolymer-indels --input Genome_minION.fasta \
--reference Genome_NCBI_REF.fasta \
--output Genome_minION_CORRECTED.fasta
Produced the following output:
OVERALL & PER-SEQUENCE STATS
===============================================
Num input sequences ..........................: 1
Num homopolymers associated with INDELs ......: 529
Num actions ..................................: 597
Num insertions ...............................: 292
Num deletions ................................: 305
* contig_332_pilon
- Homopolymers: 529
- Insertions: 292
- Deletions: 305
Corrected output FASTA .......................: Genome_minION_CORRECTED.fasta
Then copying the output file as the input file,
cp Genome_minION_CORRECTED.fasta Genome_minION.fasta
And re-running the script the same way multiple times gave the following outputs:
# (first round)
* contig_332_pilon
- Homopolymers associated with INDELs: 29
- Insertions: 21
- Deletions: 10
# (second round)
* contig_332_pilon
- Homopolymers associated with INDELs: 17
- Insertions: 9
- Deletions: 13
# (third round)
* contig_332_pilon
- Homopolymers associated with INDELs: 4
- Insertions: 4
- Deletions: 0
# (fourth round)
* contig_332_pilon
- Homopolymers associated with INDELs: 0
- Insertions: 0
- Deletions: 0
At the end, there were no more homopolymers associated with INDELs.
Please consider the following points:
-
If the input and reference genomes are not closely related enough (i.e., expected ANI if there were no sequencing errors > 98%-99%), this process may yield very incorrect outcomes. But it should work great for genomes reconstructed from the same culture.
-
The iterative improvement of a given input genome may reach to a ‘back-and-forth’ situation where there is no overall improvement, but the homopolymers associated with INDELs do not reach to 0. This happens when there are repeats in the reference genome that are identical to each other expect the number of nucleotides in homopolymers.
-
You can always add
--verboseto your command to see every single case that is considered, and resolution anvi’o reached. -
The script cleans after itself. But if you add the flag
--debugto your call, you will find the raw blast output in XML form, which is the primary file this script uses to identify and correct INDELS associated with homopolymers. -
Under all circumstances, it is important to double check your results, and make sure you keep in mind that anything you see outstanding in your downstream analyses may be due to this step.
A real-world example
This example involves two circular bacterial genomes, W01 and W48, both of which were reconstructed using minION long-reads that were assembled by Flye and polished by Pilon.
Although W01 and W48 were supposed to be near-identical genomes based on our understanding of the system, the pangenome contained a lot of gene clusters that were either found only in W01 or only in W48, which was quite unexpected. We thought that the spurious gene clusters were in-part due to frame-shifts caused by INDELs associated with random and erroneous homopolymers that influenced both genomes.
The following GIF shows three pangenomes for (1) the uncorrected genomes, (2) W01 corrected by W02 using --min-homopolymer-lenth 3 and (3) W01 corrected by W02 using --min-homopolymer-lenth 2. Please note that the sequence for W48 is unchanged throughout these steps, but it is only W01 that is modified:
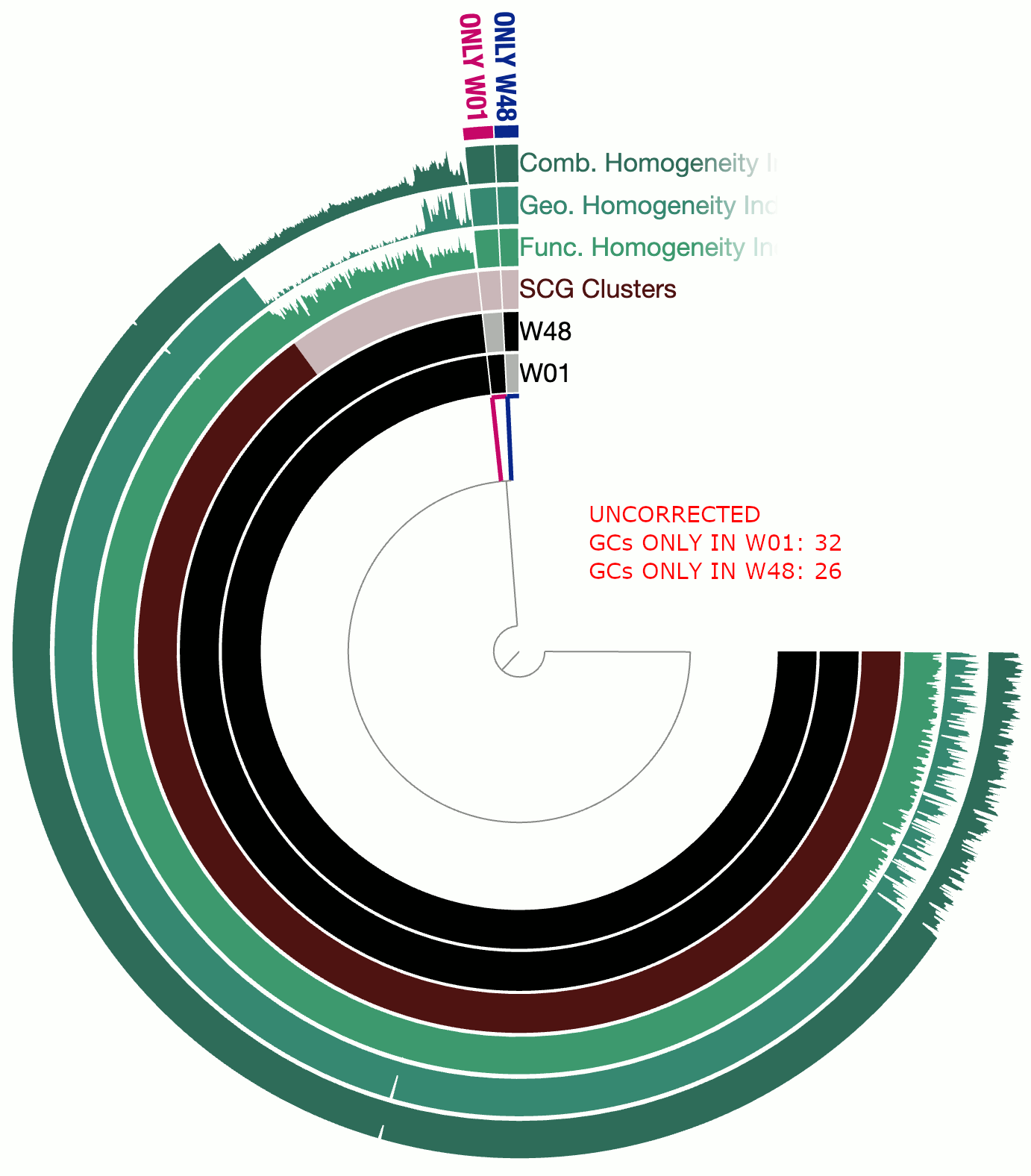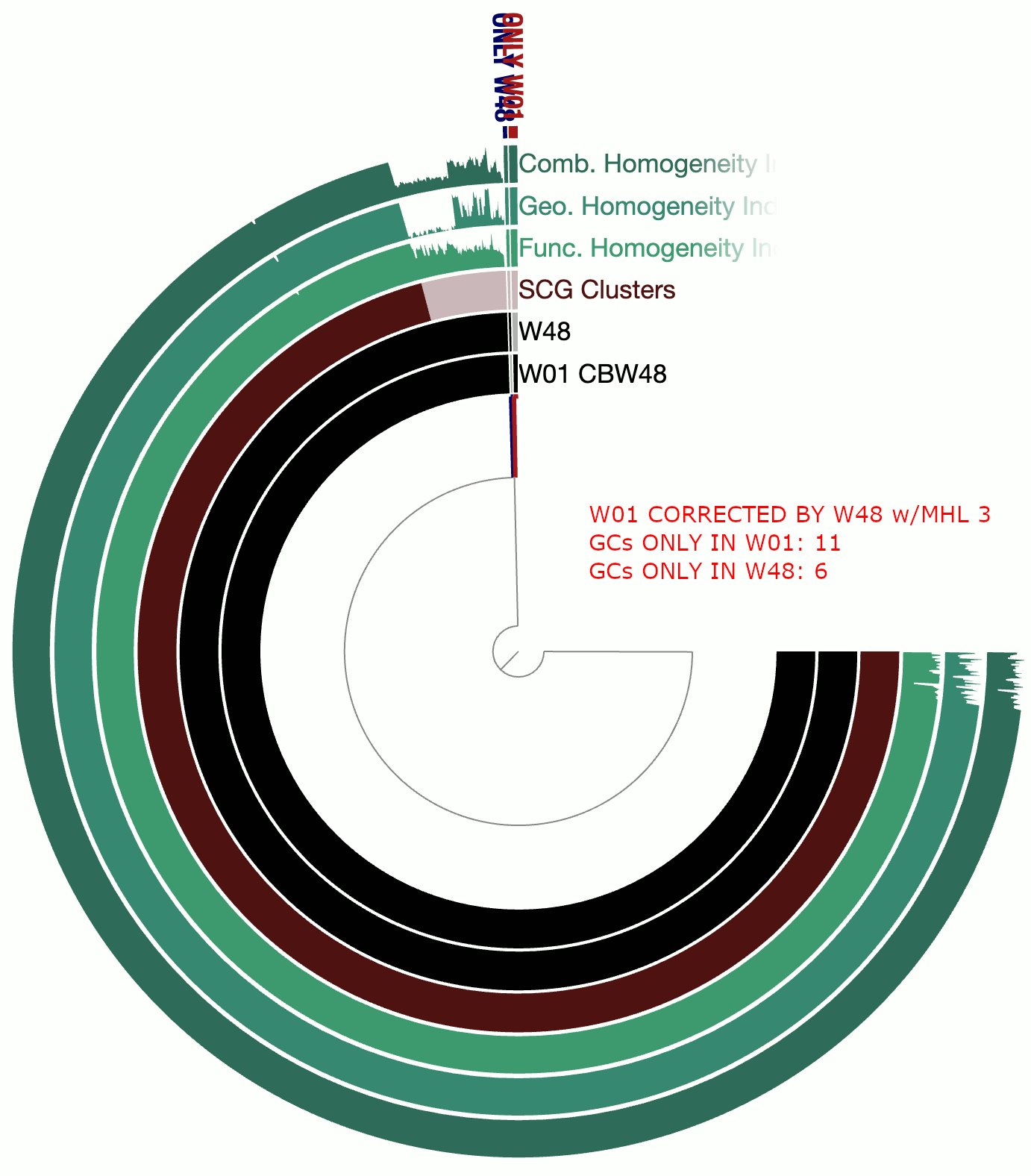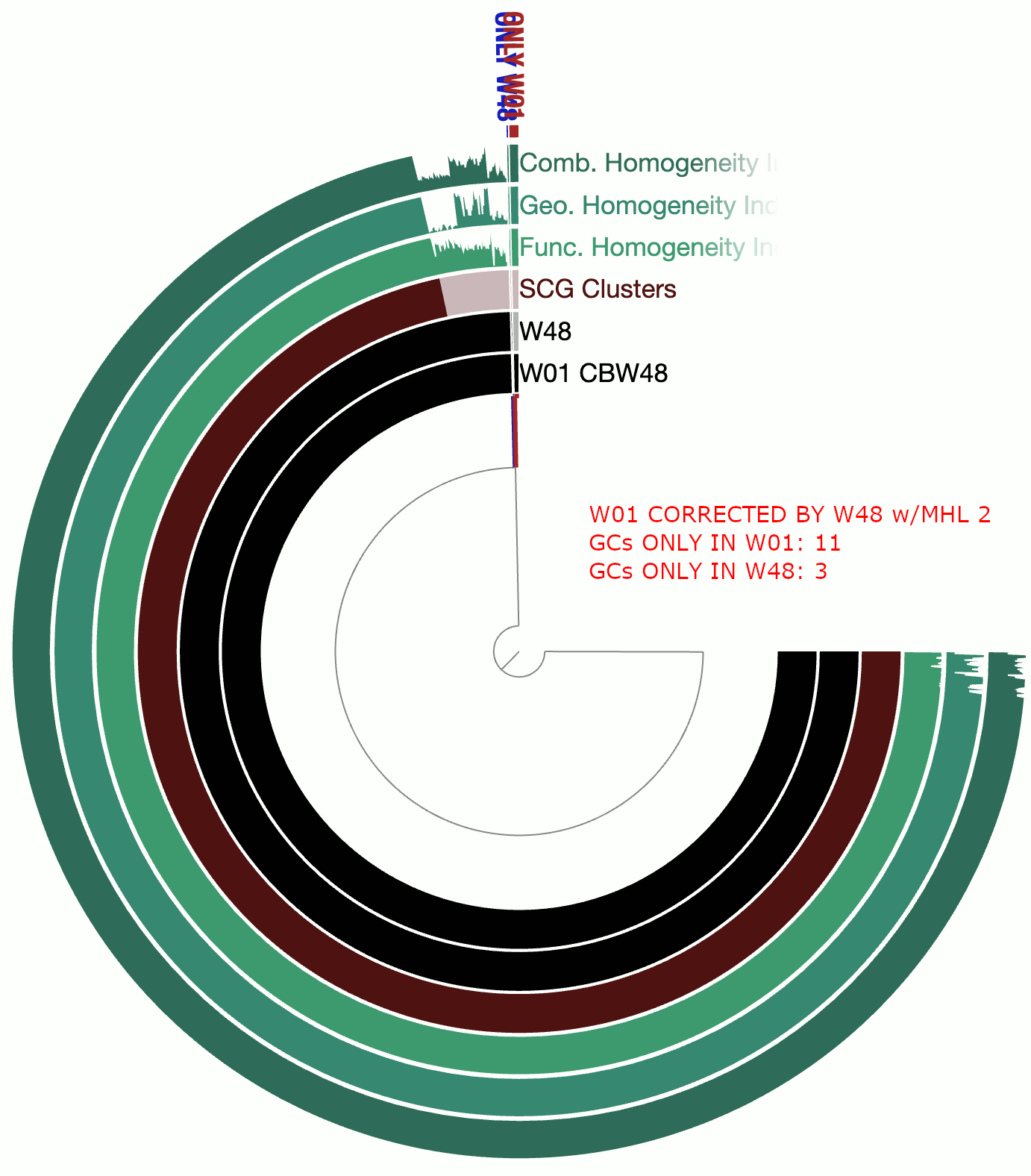
As this preliminary analysis shows, not only there is a reasonable reduction in spurious gene clusters, but also the homogeneity indices for core gene clusters display remarkable improvement. --min-homopolymer-lenth 2 seems to be doing slightly better than --min-homopolymer-lenth 3. Overall, the script seems to be doing its job.
Since in this example the ‘reference genome’, W48 is also a genome with substantial homopolymer errors, the corrected sequences in W01 do not necessarily yield amino acid sequences that are globally correct. But they are as incorrect as the ones in W48 and not more. When a very closely related reference genome is used, the corrections will not only remove spurious gene clusters from pangenomes, but also yield more accurate amino acid sequences.
For posterity, the following shell script shows how each pangenome shown in the GIF above is generated and displayed:
###########################################################################################
# NO CORRECTION
###########################################################################################
anvi-gen-contigs-database -f W01.fa -o W01.db
anvi-gen-contigs-database -f W48.fa -o W48.db
anvi-gen-genomes-storage -e external-genomes-01.txt \
-o UNCORRECTED-GENOMES.db
anvi-pan-genome -g UNCORRECTED-GENOMES.db \
-n UNCORRECTED \
--num-threads 4
anvi-display-pan -g UNCORRECTED-GENOMES.db \
-p UNCORRECTED/UNCORRECTED-PAN.db \
--title "UNCORRECTED"
###########################################################################################
# W1 CORRECTED BY W48 --min-homopolymer-length 3
###########################################################################################
anvi-script-fix-homopolymer-indels -i W01.fa \
-r W48.fa \
--min-homopolymer-length 3 \
-o W01_CBW48_MHL3.fa
anvi-gen-contigs-database -f W01_CBW48_MHL3.fa \
-o W01_CBW48_MHL3.db
anvi-gen-genomes-storage -e external-genomes-02.txt \
-o CORRECTED-BY-W48-MHL3-GENOMES.db
anvi-pan-genome -g CORRECTED-BY-W48-MHL3-GENOMES.db \
-n CORRECTED-BY-W48-MHL3 \
--num-threads 4
anvi-display-pan -g CORRECTED-BY-W48-MHL3-GENOMES.db \
-p CORRECTED-BY-W48-MHL3/CORRECTED-BY-W48-MHL3-PAN.db \
--title "W01 CORRECTED BY W48 w/MHL3"
###########################################################################################
# W1 CORRECTED BY W48 --min-homopolymer-length 2
###########################################################################################
anvi-script-fix-homopolymer-indels -i W01.fa \
-r W48.fa \
--min-homopolymer-length 2 \
-o W01_CBW48_MHL2.fa
anvi-gen-contigs-database -f W01_CBW48_MHL2.fa \
-o W01_CBW48_MHL2.db
anvi-gen-genomes-storage -e external-genomes-03.txt \
-o CORRECTED-BY-W48-MHL2-GENOMES.db
anvi-pan-genome -g CORRECTED-BY-W48-MHL2-GENOMES.db \
-n CORRECTED-BY-W48-MHL2 \
--num-threads 4
anvi-display-pan -g CORRECTED-BY-W48-MHL2-GENOMES.db \
-p CORRECTED-BY-W48-MHL2/CORRECTED-BY-W48-MHL2-PAN.db \
--title "W01 CORRECTED BY W48 w/MHL2"
Edit this file to update this information.
Additional Resources
Are you aware of resources that may help users better understand the utility of this program? Please feel free to edit this file on GitHub. If you are not sure how to do that, find the __resources__ tag in this file to see an example.