Constrained binning and the anvi’o metabins


Table of Contents
Summary
This post describes how genome-resolved metagenomics can be done manually with anvi’o even when a very large number of contigs have been assembled. Instead of processing and visualizing all contigs together, they are first clustered into self-sustained metabins using constrained binning. Each metabin can then be effectively explored to characterize and curate environmental genomes of interest. This approach has been successfully applied to multiple projects, including extra-large metagenomic co-assemblies from the Tara Oceans expeditions.
Classic genome-resolved metagenomics with anvi’o:
Regardless of their positioning in the Tree of Life, contigs from the same genome will tend to have nearly identical distribution patterns across samples in which they occur, a similar sequence composition (k-mer counts), and a complement of single copy core genes. Taking advantage of these attributes, anvi’o provides programs and an interactive interface allowing the manual characterization and curation of environmental genomes from individual metagenomic assemblies or the co-assembly of multiple metagenomes. It computes the coverage of contigs across samples, calculates their sequence composition (usually the tetranucleotide frequency), and searches for single copy core gene collections corresponding to the three domains of life. Anvi’o is now actively used by a wide range of researchers to characterize in a manual mode environmental genomes for known and unknown lineages within Bacteria, Archaea, Eukarya and viruses.
We selected three metagenomes from the DeepWater Horizon oil spill to exemplify how anvi’o can be used to characterize an environmental genome of interest with confidence (Figure 1A). A co-assembly of the three metagenomes was performed, and the anvi’o metagenomic workflow was then followed to produce the anvi’o contigs-db and profile-db files. The processed data is available for teaching or training purposes. First, the ‘anvi-interactive’ program was used to visualize the assembly in the context of relevant information (Figure 1B). In the interface, contigs were organized based on the differential coverage and tetranucleotide frequency. One could immediately identify a cluster of contigs that have a similar distribution pattern and display complementary single copy core genes indicating they represent a bacterial genome enriched in the oil plume. This cluster was selected and named “Ca. nobermanella desum”. The ‘anvi-refine’ program was subsequently used to further assess the biological relevance of this enviromnetal genome. This time, the contigs were organized based on the tetranucleotide frequency alone (Figure 1C). With the ability to visualize critical information in the interface, we could gain confidence in the quality of this environmental genome. Finally, the program ‘anvi-summarize’ was used to compute main statistics of the environmental genome for “Candidatus nobermanella desum”.
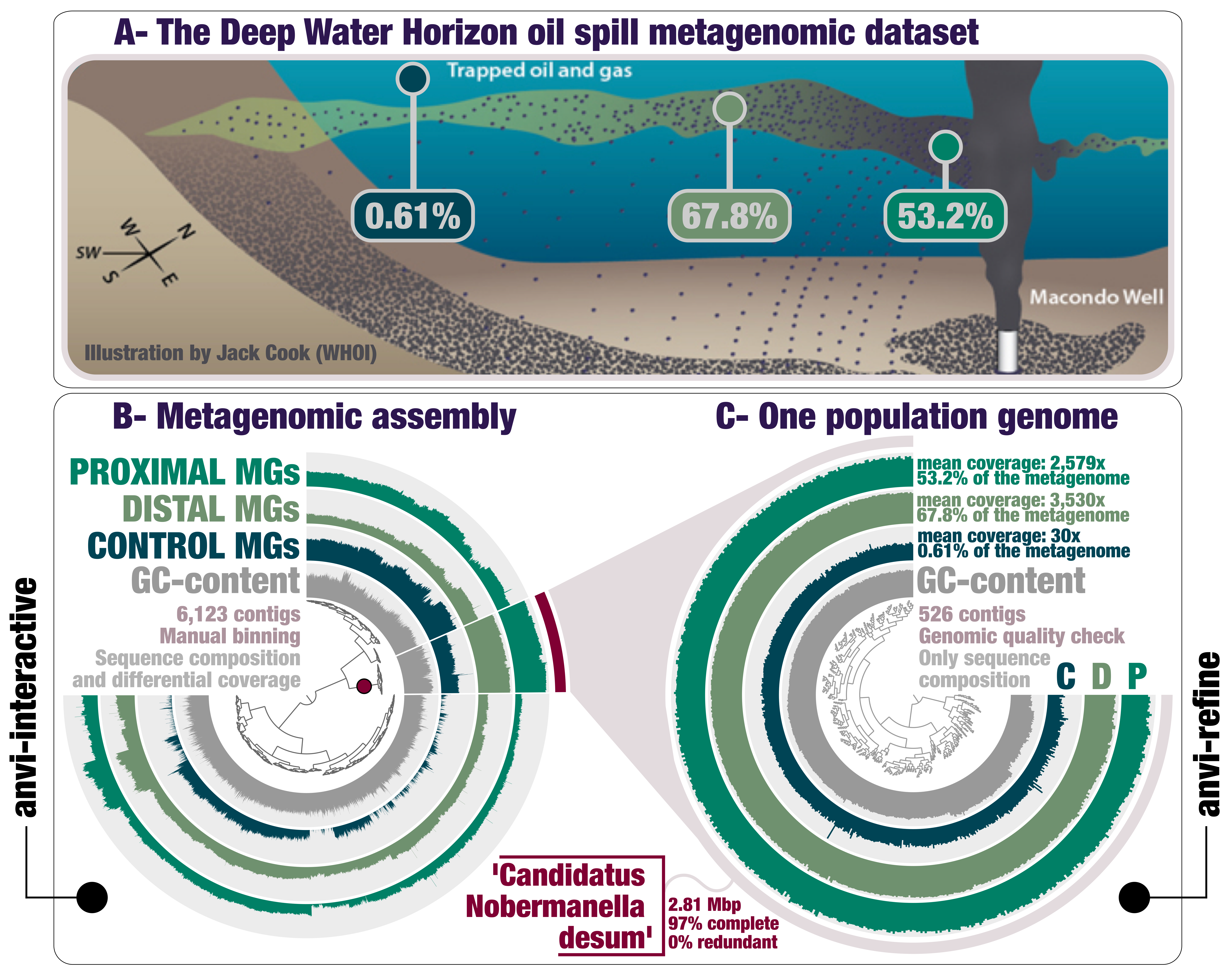
Figure 1: The anvi’o interactive interface for manual genome-resolved metagenomics. Panel A describes a metagenomic data set of 3 samples from the DeepWater Horizon oil spill (https://www.nature.com/articles/ismej201259). Panel B describes the anvi’o interactive interface visualizing the processing of 6,123 contigs assembled from the metagenomic data set. Contigs are organized based on their sequence composition and differential coverage across the 3 samples. One cluster of contigs is selected. Panel C describes the refinement of the selected cluster of contigs, which are organized based on their sequence composition alone.
Constrained genome-resolved metagenomics and the anvi’o metabins:
Anvi’o allows you to process contigs and visualize them on the interactive interface. However, it cannot cluster and visualize an infinite number of contigs. Simply, it would be too time-consuming to cluster them, and there is also a limit on the number of objects that can be visualized in the interactive interface. We usually put the limit at around 30,000 contigs to be efficiently clustered and visualized within anvi’o. In the example of the DeepWater Horizon oil spill, the assembly produced about 6,000 contigs and so it was possible to process the entire data. However, in other projects the assembly outcomes can produce much more than 30,000 contigs. In those cases, anvi’o provides an alternative solution that allows the manual characterization and curation of environmental genomes regardless of the size of your assembly: constrained genome-resolved metagenomics (a.k.a, constrained binning).
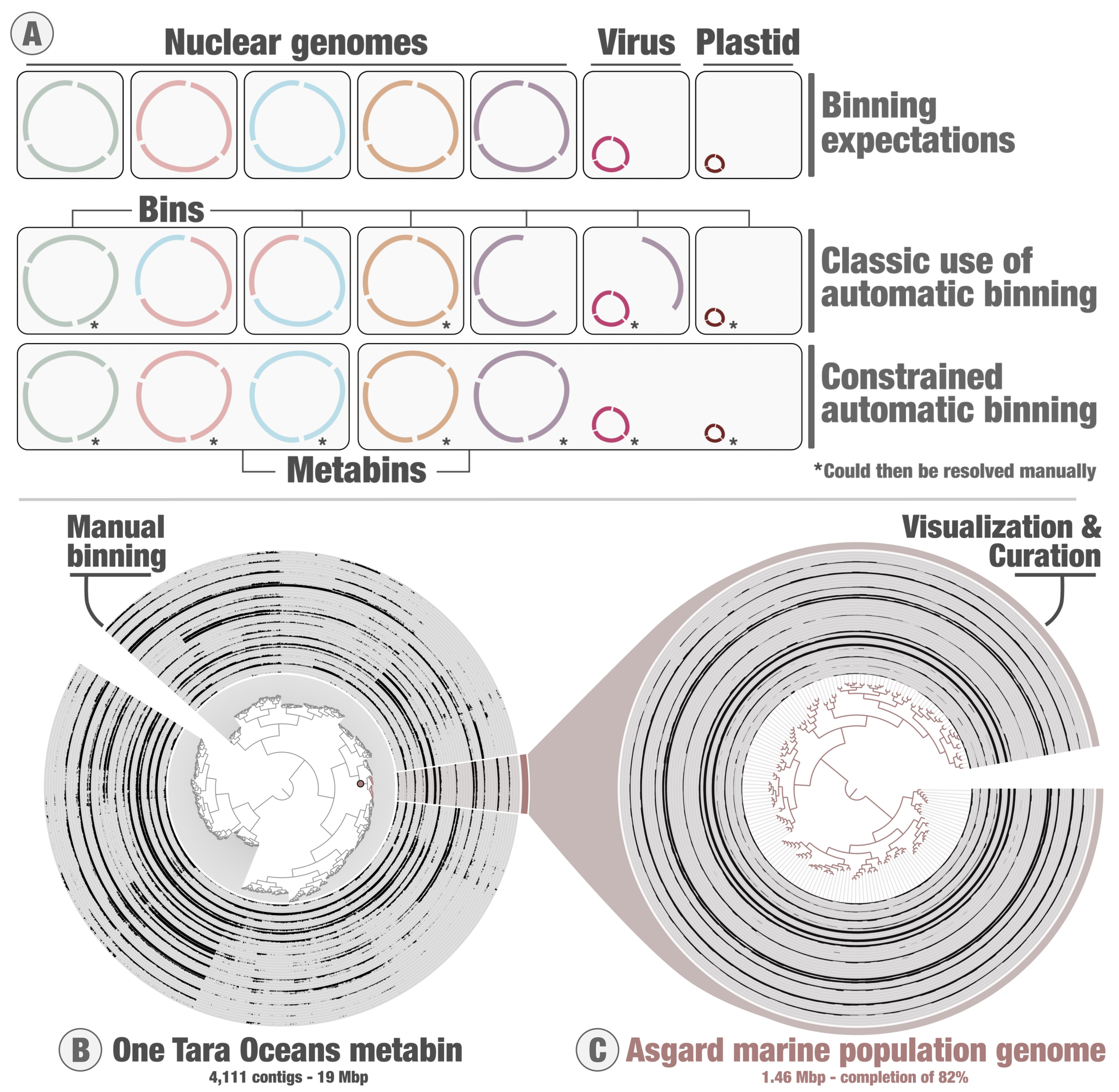
Figure 2: Constrained genome-resolved metagenomics and the anvi’o metabins. Panel A describes the concept of constrained binning as compared to the classic use of automatic binning algorithms. Panel B describes the anvi’o interactive interface for genome-resolved metagenomics, applied to one Tara Oceans metabin of ~4k contigs organized based on their differential coverage and sequence composition. Panel C corresponds to the manual curation of a subset of contigs from that metabin, which corresponds to an environmental genome of Asgard Archaea. Contigs are organized based on their sequence composition alone.
Over the years, many algorithms have been created to automatically identify environmental genomes from metagenomic assemblies. Many researchers elect to rely entirely on these automatic binning algorithms. Under this classic automatic binning framework, the algorithms try to delineate environmental genomes, which can then be incorporated into anvi’o to visualize and curate them. However, automatic binning can place contigs from the same environmental genome in different bins (fragmentation error). This is where constrained binning comes in. Instead of going after individual environmental genomes, the approach aims at generating a defined number of metabins that by design can include multiple environmental genomes that are often taxonomically unrelated (see concept in Figure 2A). The CONCOCT algorithm, which can be run from within anvi’o with the program ‘anvi-cluster-contigs’, has a flag to constrain binning to a defined set of clusters. If you ask for 100 clusters, the algorithm will produce 100 metabins. The metabins are stored as a collection in the anvi’o PROFILE database, and it is then possible to generate self-sustained metabins using the ‘anvi-split’ program. This way, each metabin can be explored and shared individually. The first key advantage of constrained binning is that contigs from the same environmental genome will rarely end up in different metabins (Figure 2A). The second key advantage is that each metabin can be processed and visualized with anvi’o for manual binning (Figure 2B) and curation (Figure 2C). The Figure 2 shows the example of one Tara Oceans metabin from the Indian Oceanic. This metabin contains the environmental genome of an Asgard Archaea.The processed data is available for teaching or training purposes. The third key advantage is that if you are looking for something specific (e.g., taxonomic signal for your favorite lineage or function), anvi’o can help you identify the metabins that require your attention. This way, you can effectively go after any signal of interest even in the case of very large assemblies. If you are curious about targeted binning in the context of constrained binning, this blog tells the story of how phylogeny-guided genome-resolved metagenomics was used to identify anvi’o metabins containing environmental genomes for a previosuly unnoticed virus phylum.
How to define the number of metabins:
Chosing the number of metabins depends on the number of contigs beeing processed, but might also be influenced by the number of distinct genomes present within the assembly. The anvi’o program ‘anvi-display-contig-stats’ computes the frequency of each single copy core gene from the 3 domains of life to provide an estimated number of assembled microbial genomes. These frequencies provide a reasonable proxy for the genomic complexity of metagenomic assemblies but for now it does not take into account virus genomes.
Ultimately, the determination of the number of metabins is your’se to make depending on your project and aims. But as a perspective, we share choices made for nearly 1000 Tara Oceans metagenomes. 11 large metagenomic co-assemblies were performed one oceanic region at a time. For instance, metagenomes from the Mediterranean Sea were co-assembled together, providing nearly 2 million contigs >2.5 kb in length. A total of 400 metabins were generated from this co-assembly, based solely on the number of contigs. For smaller assemblies (e.g., from the Red Sea), a smaller number of metabins was set. Overall, in that project there were around 5,000 contigs per metabin, in average (Figure 3). It is important to keep in mind that the metabins can widely vary not only in size, but also in biological value. Some metabins might contain various high-quality environmental genomes (often with high N50 scores) while others will only contain noise we currently fail to make sense of (often with low N50 scores).
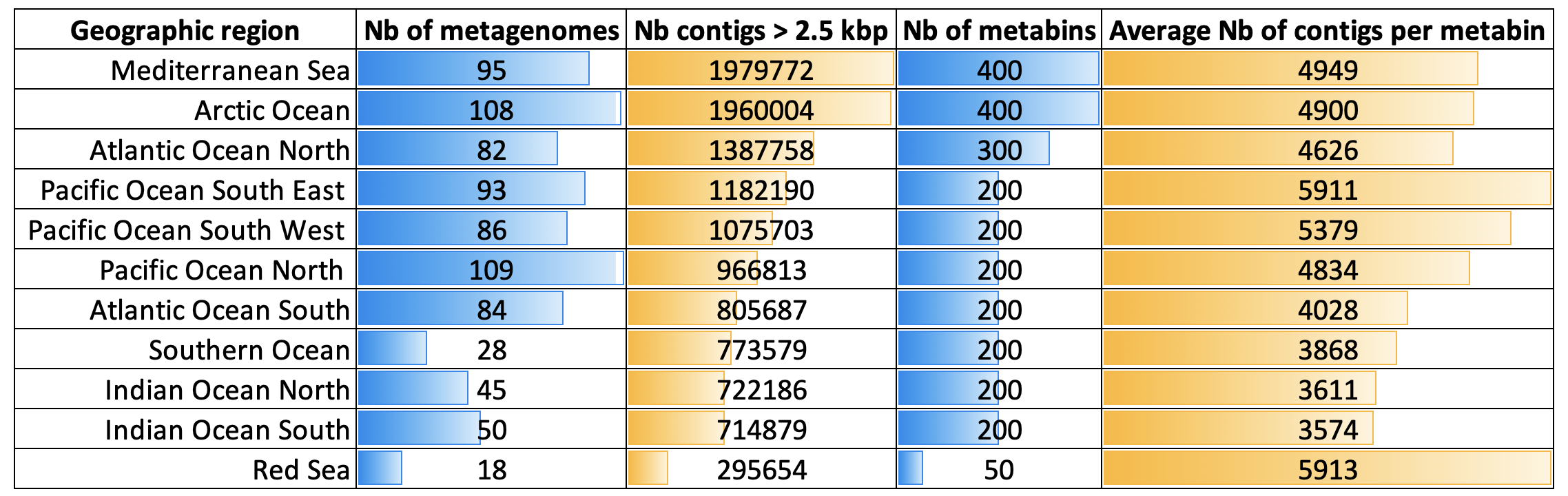
Figure 3: The number of metabin for each large Tara Oceans metagenomic co-assembly. The figure summarizes the metagenomic co-assembly outcomes for 11 large Tara Oceans metagenomic co-assemblies, and the number of metabins that were generated from this paper.
Examples of metagenomic surveys that relied on constrained binning:
Constrained binning has already been used to manually screen the generated metabins and extract environmental genomes from large metagenomic co-assemblies corresponding to both environmental and human-related ecosystems (e.g., the oceans, oral cavity, or else radioactive waste disponal). The detailed bioinformatic workflow for the creation of anvi’o metabins from Tara Oceans metagenomic co-assemblies can give you extra guidance. In addition, constrained binning has also been used to perform targeted binning, where only a subset of metabins containing signal of interest is explored (e.g., in the oceans or for mosquitoes).