anvi-reorient-genomes
Table of Contents
Reorient circular genomes and scaffold fragmented genomes in a fasta-txt so their coordinates match a chosen reference genome using minimap2 alignment and seqkit manipulation..
🔙 To the main page of anvi’o programs and artifacts.
Authors


Can consume
Can provide
Usage
anvi-reorient-genomes aligns genomes listed in a fasta-txt to a chosen reference genome and reorients them to match the reference coordinate system. For circular genomes (complete bacterial genomes, plasmids, viral genomes), it rotates and/or reverse-complements them to synchronize their arbitrary circularization beginnings with the reference. For fragmented genomes (MAGs, draft assemblies), it orders and orients contigs to match the reference genome’s coordinate system. By doing so, it maximizes gene order conservation across genomes, so that downstream analyses that rely on synteny can have an easier time.
It reports (1) alignment quality between each pair of genomes (coverage between the two genomes and very crappy approximate ANI that no one should trust), (2) what actions were taken to reach a consensus (reverse-complement, rotations, contig ordering, etc), and (3) a final determination of the level of trust one should have for outcomes. The program also generates synteny ribbon plots to visualize the alignment patterns between each genome and the reference before and after reorientation.
The default usage is simple, which will simply instruct anvi’o to take care of everything de novo,
anvi-reorient-genomes --fasta-txt fasta-txt \ --output-dir REORIENTED-FASTA-FILES/
Another option is to tell anvi’o to use a specific genome as reference to to orient all others:
anvi-reorient-genomes --fasta-txt fasta-txt \ --reference REF-GENOME-NAME \ --output-dir REORIENTED-FASTA-FILES/
Or one can ask anvi’o to first use the DnaA gene to orient the reference before orienting all others:
anvi-reorient-genomes --fasta-txt fasta-txt \ --use-dnaa-for-reference-orientation \ --output-dir REORIENTED-FASTA-FILES/
De novo identification of reference orientation
TL;DR: Best for any set of highly similar genomes - whether circular (viral, plasmids, complete bacterial genomes) or fragmented (MAGs, draft assemblies). For circular genomes, uses secondary alignments from minimap2 to find the most conserved position across all genomes in a data-driven manner. For fragmented genomes, orders and orients contigs based on their alignment to the reference.
If the user does not explicitly mention a reference genome, the program will auto-select the genome with the fewest contigs as reference (ties broken by longest total length), preferring complete circular genomes over fragmented ones. It will then rotate the reference genome to an optimal starting position that is conserved across most of the input genomes mentioned in the fasta-txt file in a mindful fashion. It is ‘mindful’ because anvi-reorient-genomes does this step by first aligning each genome to the reference using all possible good alignments (primary + secondary alignments) to get a true picture of conserved regions across all genomes, and then using a 1,000 bp sliding window to search for a region that is shared between all genomes in a greedy fashion (once it finds a region that works for all genomes, it stops).
Please note that when a reference genome is chosen automatically, the program will likely end up rotating the automatically chosen reference genome to a more meaningful and conserved start position to maximize agreements across all genomes.
If you have a reference genome that you trust (downloaded from a reliable source or manually circularized), you can ask the program to use that as a reference. In which case the program will not be tinkering with the reference, and do its best to match every other genome to it.
Using DnaA gene for biologically meaningful reference orientation
TL;DR: Best for any highly similar set of bacterial genomes. If DnaA is not found in the reference, the program will issue a warning and proceed without rotating the reference.
For bacterial genomes, you can use the --use-dnaa-for-reference-orientation flag to orient the reference genome based on the DnaA gene, which typically marks the origin of replication:
anvi-reorient-genomes --fasta-txt fasta-txt \ --use-dnaa-for-reference-orientation \ --output-dir REORIENTED-FASTA-FILES/
This approach will use prodigal to call genes in the reference genome, use hmmsearch with the Bac_DnaA_C HMM profile from Pfam to identify the DnaA gene, rotate the reference to start at the DnaA gene position, then align all other genomes to this DnaA-oriented reference.
For the most up-to-date list of parameters and their default values, please see the help menu on your terminal.
Example terminal output
Outputs in the terminal will help you figure out what is going on, so please keep an eye on them.
Here is an example output for two circular genomes where the query nicely aligned to the reference (the alignment plots always show before and after the alignment):
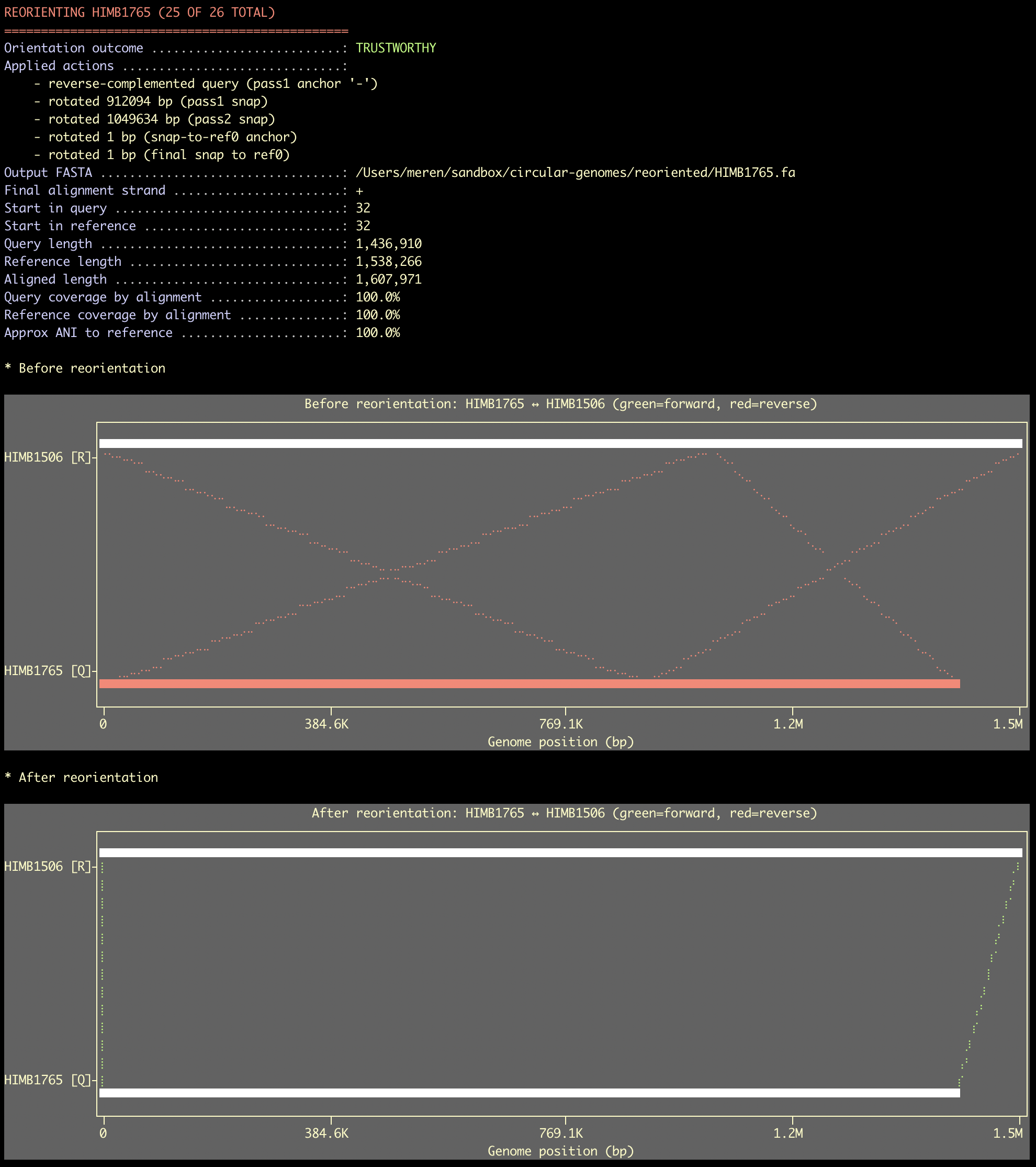
The fact that ‘Start in query’ and ‘Start in reference’ is identical is great news, and shows that the program was able to set a start position that works perfectly. The high coverage alignment between the two is also great as it shows that you brough together genomes that are quite closely related. The approximate ANI says 100%, and it is not to be trusted. For instance, the true ANI between these two genomes in this example is about 93%. But since we are cutting corners using the minimap2 aligned bits between the two, we really really overestimate the ANI value. It is useful, but please don’t rely on the apprximate ANI reported here for anything serious.
Here is another example for two circular genomes where the query did not aligned well to the reference, which means, the reorientation is highly unreliable:

The program will give you something to read and think about at the very end:
(...)
FINAL REPORT
===============================================
Your genome reorientation task considered 5 genomes and 1 reference. Some
outcomes are not trustworthy (low alignment coverage can lead to unreliable
orientation). Please review the alignment plots above and the summary below to
decide which FASTA files to use downstream.
Trustworthy ..................................: 1
- d -> /Users/meren/sandbox/miserable-genomes/reoriented/d.fa
Somewhat OK ..................................: 1
- g -> /Users/meren/sandbox/miserable-genomes/reoriented/g.fa
Not trustworthy ..............................: 4
- c -> /Users/meren/sandbox/miserable-genomes/reoriented/c.fa
- b -> /Users/meren/sandbox/miserable-genomes/reoriented/b.fa
- f -> /Users/meren/sandbox/miserable-genomes/reoriented/f.fa
- m -> /Users/meren/sandbox/miserable-genomes/reoriented/m.fa
Failed .......................................: 0
✓ reorient_genomes.py took 0:00:15.245738
Here is a final example of re-orienting contigs in a fragmented genome using a reference:
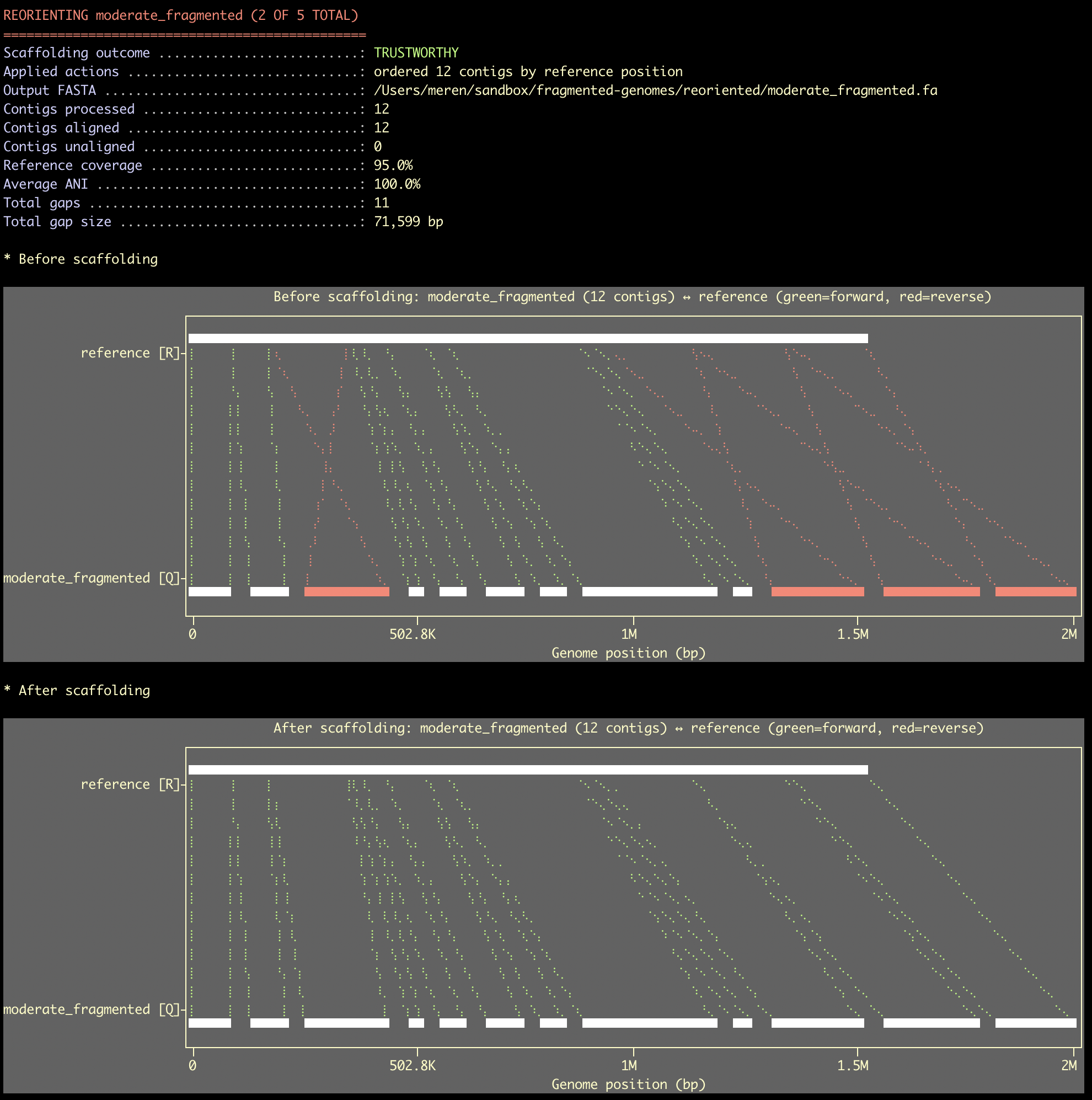
Where each contig was nicely turned around, and ordered in the final FASTA file to follow the right order that matches to the genomic context of the reference.
If you wanted, you could use the flag --scaffold-fragmented and make anvi’o produce a single contig FASTA file where ‘missing’ content in the query genome is filled with N bases to create your own ‘complete genome’ to prank your microbiologist colleagues:

Please don’t do it, though – don’t ‘scaffold’ your MAGs based on a reference and then submit them to databases as single-contig genomes. Procedure above is excellent to have gene synteny preserved despite the missing content, but that is about it.
What the program does
Here is a more detailed description of what is going on behind the scenes when you press ENTER:
-
Parse inputs and pick a reference. Reads fasta-txt, and if
--referenceis not set, the program picks the genome with the fewest contigs (ties broken by longest total length). All FASTAs are sanity-checked (existence, FASTA format). The reference must be a single-contig circular genome. -
Determine reference orientation. The program uses one of three strategies to orient the reference genome:
- DnaA-based orientation (if
--use-dnaa-for-reference-orientationis set): Calls genes withprodigal, searches for the DnaA gene usinghmmsearchwith the Bac_DnaA_C HMM profile, and rotates the reference to start at the DnaA gene position. This provides biologically meaningful orientation for bacterial genomes. - De novo optimal position (if reference is auto-selected without DnaA flag): Aligns each genome to the reference using minimap2 with
--secondary=yes -N 100 -p 0.5to capture all possible good alignments (not just the best one). Builds a coverage map using 1,000 bp bins showing which positions are covered by alignments from each genome. Identifies the position with maximum coverage across all genomes (stopping early if it finds 100% coverage). The reference is then rotated to start at this optimal position, ensuring all genomes will start at a conserved region that is genuinely shared across the dataset. - User-specified reference (if
--referenceis set): Uses the reference genome as-is without rotation.
- DnaA-based orientation (if
For circular genomes (single-contig):
-
Initial alignment (reference vs query). Runs
minimap2(presetasm5, with as many threads as you asked for using--threads) to align each query to the reference and identifies a primary anchor near reference position 0. If the anchor is on-strand, the query is reverse-complemented. The query is rotated so that reference position 0 maps onto the query (first snap). -
Second alignment and snap. Re-aligns the rotated query, finds the primary anchor with the smallest reference start, and rotates again to bring reference 0 onto the query (second snap).
-
Snap-to-zero with a ref0-focused anchor. Aligns once again, picks the primary anchor closest to reference position 0, rotates, aligns once more, and applies a final snap so that reference position 0 maps to query position 0.
-
Iterative correction for perfect alignment. After the final snap, the program checks if the alignment truly starts at position 0 in both the query and reference. If not (e.g., due to
minimap2soft-clipping divergent regions), it calculates the necessary rotation, applies it, and re-aligns. This iterates up to 5 times or until the genomes are perfectly aligned at position 0.
For fragmented genomes (multi-contig MAGs or draft assemblies):
-
Contig filtering. Contigs shorter than
--min-contig-length(default: 1,000 bp) are excluded from processing. -
Individual contig alignment. Each contig is independently aligned to the reference using
minimap2. The program detects contigs that span the circular boundary of the reference (wrap-around contigs) and splits them into separate parts for proper ordering. -
Contig ordering and orientation. Contigs are sorted by their alignment position on the reference genome. Contigs aligned to the reverse strand are reverse-complemented to match the reference orientation.
-
Output generation. By default, contigs are written as separate sequences in the output FASTA, ordered and oriented to match the reference. If
--scaffold-fragmentedis used, contigs are concatenated into a single sequence with N-padding representing gaps based on the reference genome distances.
For all genomes:
-
Write outputs. Copies the reference fasta to the output directory (potentially rotated if auto-selected). Writes each reoriented query fasta under the same name (and using the original extension).
-
Report per-genome stats and alignment plots. For each genome, the program reports the orientation outcome (whether it was
TRUSTWORTHY,SOMEWHAT OK, orNOT TRUSTWORTHYbased on alignment coverage), many other statistics, and synteny ribbon plots showing the alignment patterns before and after reorientation to visualize orientation quality. -
Final report. Summarizes the number of genomes in different trust categories along their output FASTA paths for the user to decide which outputs are safe for downstream analyses.
Tips, caveats, and runtime
-
Interpreting “Start in query” and “Start in reference” (circular genomes only): These numbers show where
minimap2primary alignments begin. When both values are equal (e.g., both 61), your genomes are correctly aligned at the same biological position. The small offset is just natural sequence variation in the first few dozen base pairs sinceminimap2soft-clips divergent regions at the beginning. In fact, Meren was so concerned about it, he triple-checked this, and discovered that despite differences in offsets across genomes, the amino acid sequences of the first genes in every aligned genome was identical even though their ‘start’ positions in individual genomes differed. So that’s that. But when the values differ (e.g., query=0, reference=106), the alignment may have issues and the genome might not be well-oriented. -
Interpreting trust labels: For circular genomes, low coverage (either genome covered <50%) yields
NOT TRUSTWORTHY. For fragmented genomes, the label considers both reference coverage and the percentage of contigs that aligned. This often means the genome is too divergent or structurally different to reorient confidently. Check the alignment plots and stats before using such outputs. Genomes withTRUSTWORTHYlabels are safe for downstream comparative analyses. -
Understanding alignment visualizations: The synteny ribbon plots show alignment blocks as ribbons connecting the reference (top) to the query (bottom). Green ribbons indicate forward-strand alignments, red ribbons indicate reverse-strand alignments. For fragmented genomes in the “before” plot, each contig appears as a separate segment with gaps between them. After reorientation, contigs are ordered and oriented to match the reference. Contig segments in the query genome are color-coded: white for forward-strand contigs, red for reverse-complemented contigs, and green for contigs that were split due to wrapping around the circular reference boundary.
-
Scaffolding fragmented genomes: By default, fragmented genomes are written with contigs as separate sequences (ordered and oriented). Use
--scaffold-fragmentedto concatenate them into a single sequence with N-padding representing gaps. While this maximizes gene synteny for comparative analyses, do not submit N-scaffolded MAGs as complete genomes to public databases. The scaffolding is reference-based and may not represent the true genomic structure. -
Auto-selected reference and optimal start: When you don’t specify
--reference, the program picks the genome with the fewest contigs (ties broken by longest total length), preferring complete circular genomes over fragmented ones. It then rotates the reference to a conserved position across your dataset using secondary alignments to capture all conserved regions (not just the single best alignment). This is especially useful for sets of closely related genomes where you want them all to start at a biologically meaningful position. For bacterial genomes, consider using--use-dnaa-for-reference-orientationfor even more consistent results based on the replication origin. If you want a specific genome or starting position, use--referenceto override this behavior. -
DnaA-based orientation benefits: When working with bacterial genomes, the
--use-dnaa-for-reference-orientationflag typically produces highly consistent alignments (e.g., all genomes starting within a few base pairs of each other) because it uses biological knowledge (the replication origin) rather than purely sequence-based heuristics. This can be particularly valuable for downstream synteny analyses or when comparing gene order across closely related strains. -
Approximate ANI: This is quite a garbage number at this point, and it is no one’s fault. When
minimap2emitsdv, ANI is computed as(1 - dv) x 100. Otherwise it falls back tonmatch/alen x 100. So treat it as a quick proxy, not an accurate ANI calculation. It is not the purpose here. -
Circular ambiguity: Circular genomes can align equally well at different offsets. The program applies multiple snaps and iterative corrections to align reference position 0 to query position 0, but in highly repetitive cases the true biological origin may still be ambiguous.
-
Visualization options: By default, alignment plots are generated to help you assess the quality of reorientation. Use
--skip-visualizing-alignmentsto disable plotting for faster processing when you only need the FASTA files. Customize plot dimensions with--plot-widthand--plot-height(note: widths below 100 characters may not display properly). -
Runtime: Each circular genome triggers several
minimap2runs andseqkitrotations. Fragmented genomes are faster as each contig is aligned only once. The optimal start finding step (when auto-selecting reference) adds an initial survey phase that uses secondary alignments for more accurate conserved region detection. The--use-dnaa-for-reference-orientationflag adds gene calling and HMM search overhead (a few seconds for a typical bacterial genome). Overall, it takes no more than 30 seconds on a laptop computer to reorient 30 SAR11 genomes using the de novo approach, and slightly longer with DnaA-based orientation.
Edit this file to update this information.
Additional Resources
Are you aware of resources that may help users better understand the utility of this program? Please feel free to edit this file on GitHub. If you are not sure how to do that, find the __resources__ tag in this file to see an example.