anvi-report-circularity
Table of Contents
Predict contig circularity from paired-end read alignments in a given BAM file. This program samples insert sizes, looks for RF pairs spanning junctions, and reports per-contig circularity statistics..
🔙 To the main page of anvi’o programs and artifacts.
Authors

Requires
Provides
Usage
This program takes advantage of paired-end read technology and uses reverse-forward (RF) read pairs to determine whether a given contig in a given bam-file is circular, linear, or indeterminate by making use of the insert size distributions in the entirety of the bam-file.
The default command-line usage is simple:
and here is a real example:
anvi-report-circularity AP7F060721I7.bam \ -o AP7F060721I7-REPORT.txt
But there are a number of parameters that can be tuned to adjust to the dataset. For the most up-to-date list of parameters and their default values, please see the help menu on your terminal.
Some background
Read pairs from Illumina-style paired-end sequencing map back to the DNA template from which they originate in the Forward-Reverse orientation (FR) since the sequencer essentially reads towards the center of the DNA molecule from its both ends. The ‘insert size’, i.e., the total size of the DNA molecule is determined by library preparation prior to sequencing, often has a relatively tight distribution after a careful size selection step (which is not very common, but always a good idea since a relatively uniform size distribution also help downstream assembly tasks).
You can always see a histogram of insert sizes in your bam-file using anvi-get-tlen-dist-from-bam and figure out how well you did.
While FR orientation is what is expected for the vast majority of paired-end sequences, when a circular DNA molecule ends up being linearized during assembly, read pairs that originally spanned the circularization point now map back to the linear DNA as Reverse-Forward (RF) at opposite ends of a given contig. Then, if a given contig has a lot of RF reads on it, one can claim that the contig was circular in the sample if the ‘circular insert size’ of these RF pairs at its edges match the overall insert size distribution of the library.
This is kind of a general knowledge in the field and it is exploited by many algorithms to talk about circularity, inversions, INDELs, and all sorts of other structural variants that can be discovered by taking advantage of such oddities in mapping results. So it is difficult to pinpoint a paper to be the origin of this idea, but the paper by Ken Chen and colleageues have, BreakDancer: an algorithm for high-resolution mapping of genomic structural variation, has a very nice summary figure that explains the situation, and explains why RF pairs indicate structural junctions:
That said, anvi-report-circularity takes advantage of another situation that is not covered in this figure, which was better described in the paper “Diverse plasmid systems and their ecology across human gut metagenomes revealed by PlasX and MobMess” by Mike Yu and colleagues:
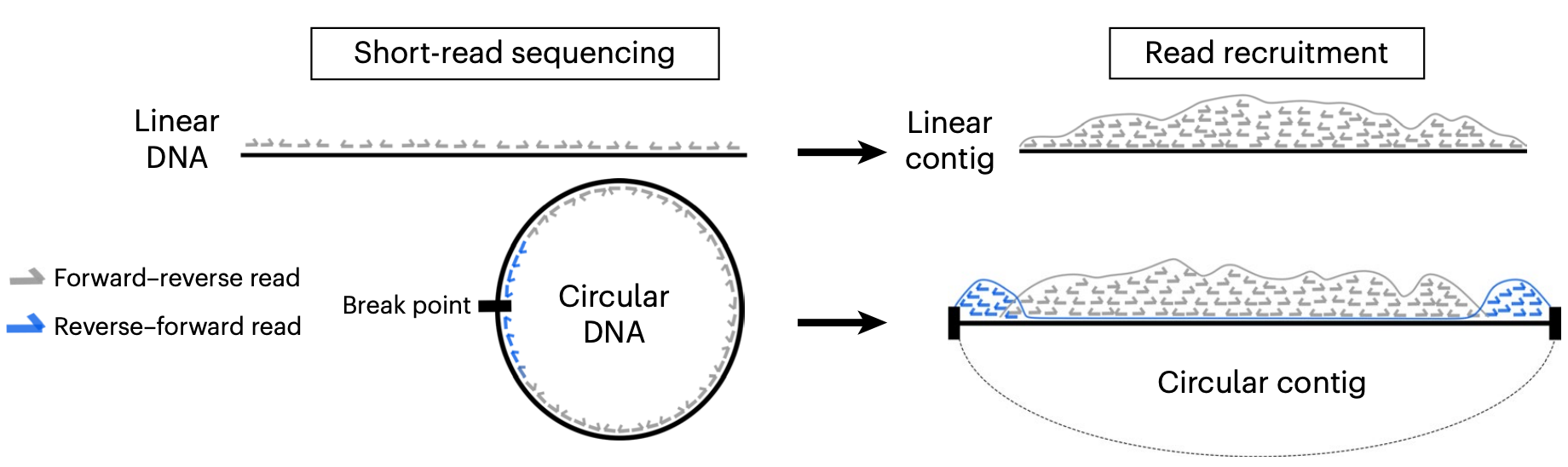
Where they used this circularity principle to assess circularity of de novo identified plasmids in metagenomes. anvi-report-circularity follows on the footsteps of that work, but improves it in important ways, and prevents the interplay between the lenght of a contig and the threshold for median insert size expectation to yield false positives for very short sequences.
We thank Sergio George Carreño, a Professor at the University of Chile who has been studying human gut plasmids at the University Medical Center Groningen, with his help with identifying these issues, and the time he put into testing anvi-report-circularity.
How it works
The following describes what anvi-report-circularity does step by step while shedding light on the purpose and relevance of the command-line parameters.
- Estimate insert-size statistics. The run starts by trying to establish an understanding of the insert size statistics by sampling up to
--max-num-pairs-for-is-estforward-reverse (FR) pairs from across all contigs (this is to make sure the program does not process ALL reads in a bam-file). Then, it computes the median insert size for the library,M, and its median absolute deviation,MAD. For this to work, a minimum of--min-pairs-for-statsFRpairs is required, otherwise we can’t have anMand so the program stops with an error already.MADshould NEVER be zero (since it would indicate that you have a hyper-uniform library, but it is not possible), but in case you are running the program with simulated data, the program simply assumesMADequals 1.0 and moves on.
If you prepared your genomic or shotgun metagenomic libraries without a proper size selection step, this program will unlikely work well since M will be irrelevant for most pairs, and MAD will be relatively useless. You can have an idea about your insert size distribution using anvi-get-tlen-dist-from-bam.
-
Scan each contig. Then the program proceeds to read in the bam-file for each contig to count
FRpairs, and keep track of reads near contig edges. If the length of a contig,Lis shorter than2 × M, they will be ignore at this step as their miserable length would make it impossible for the downstream code to confidently determine if they are circular or not. These contigs are marked asindeterminatein the output file with a warning flag. -
Compute the expected number of RFs. The algorithm then calculates the expected number of
RFpairs for a given contig of lengthLto be circular using the following logic,E[RF] = N_FR × M / (L - M)where
N_FRis the number ofFRpairs on the contig. This reflects the chance that anRFpair spans a circular junction when linearized.
The efficacy of the step above heavily depends on your assembler’s restraint to not add stupid extras such as non-existent tandem repeats to contig ends (as they generally do with circular entities) :/
-
Score RFs that support circularity. For each
RFpair, the algorithm then computes the ‘circular insert size’, let’s call itcircular_insert. Circular insert size essentially is a new insert size value calculated by the formula:left_end + (L - right_start)and simply answers the question, “*what the insert size of this
RFpaired end would have been if the contig was indeed circular in the environment?”. A givenRFis one that ‘supports’ circularity, if|circular_insert - M| ≤ T, whereTis tolerance, and it is calculated usingMADand the user-defined tolerance factor:T = --insert-tolerance-factor × MAD. Once allRFparis are considered, the overall circularity support for a given contig is the fraction of observedRFpairs that are supporting.
This step will most likely miss pro-viruses that occur in high-coverage chromosomes and found as circular genomes in capsids.
-
Assess edge coherence. This is not immediately relevant to the circularity calculation, but it is something good to have in the report: the proportion of paired-end reads at the extremeties of contigs where both mates map into the contig. If both mates in every single paired-end read that occur at the contig edges, which is defined by those that are within
Mnucleotides form either of the contig edges, the edge coherence is maximum (which may indicate that a given contig is linear, and not a fragment of anything larger). If there is a large number of paired-end reads in these edges with their mate mapping to a different contig or not mapping at all, then the edge coherence is minimum (which may indicate that the contig is a fragment of something larger). -
Report. The program concludes by generating an output file that lists for each contig their status, support scores, coverage counts, and warning flags.
Details of the decision making
The program makes use of a combination of user-defined thresholds and the min_required variable which is equivalent to --min-supporting-pairs or --expected-fraction-threshold × E[RF], whichever is larger.
Given all these, the final decision for contig status is made based on the rules below, which are evaluated in this order:
- Circular if supporting
RFpairs ≥min_requiredand circularity support ≥--circularity-support-threshold. - Circular if supporting
RFpairs ≥--min-supporting-pairsand ≥80% of observedRFpairs are supporting. - Linear if no
RFpairs are observed and there are at least 100FRpairs on the contig. - Linear if supporting
RFpairs <min_requiredand circularity support <--circularity-support-threshold. - Otherwise indeterminate (flagged as ambiguous evidence). Contigs with no
FRpairs, very short lengths (L < 2M), or extremely low edge coverage are also reported as indeterminate with somewhat informative flags.
Edit this file to update this information.
Additional Resources
Are you aware of resources that may help users better understand the utility of this program? Please feel free to edit this file on GitHub. If you are not sure how to do that, find the __resources__ tag in this file to see an example.