anvi-report-inversions
Table of Contents
- Authors
- Requires
- Provides
- Usage
- Anvi’o philosophy to find inversions
- Before you run this program
- Other essential inputs to run this program
- Identifying regions of interest
- Finding palindromes
- Confirming inversions
- Computing inversion activity
- Computing inversion activity using previously computed inversions
- Reporting genomic context around inversions
- Searching for conserved motifs in inverted-repeats
- Targeted search
- HTML output
- Other outputs
- Additional Resources
Reports inversions.
🔙 To the main page of anvi’o programs and artifacts.
Authors



Requires
Provides
Usage
This program allows you to find genomic inversions using metagenomic read recruitment results, and their activity patterns across samples.
Specifically, this program search for site-specific inversions which are carried out by site-specific recombinases. This enzyme recognizes a pair of inverted repeats (IRs). The IRs are distant from each other and the site-specific recombinase invert the DNA fragment between the IRs.
In brief, anvi’o leverages paired-read orientation (through the --fetch-filter mechanism in anvi-profile explained below) to locate regions of interest in a set of contigs. It screens for IRs within regions that are enriched in read pairs that are enriched in forward/forward or reverse/reverse orientations, and uses short-reads to confirm which IRs correspond to real inversions. Anvi’o can also compute the ‘inversion activity’, i.e., the relative proportion of each orientation of an inversion in each sample.
Anvi’o philosophy to find inversions
Much like a T-Rex, the vision of anvi’o relies on movement and it cannot see an inversion if it does not move, or in this case, invert. So let’s start with what you cannot do with this command: you cannot find inversions in a set of contigs alone.
To find an inversion, you need to have short-reads from at least one sample. If there is even a small fraction of the members of a microbial population that have an inverted sequence, then anvi-report-inversions will very likely find it for you!
Before you run this program
Anvi’o is able to locate inversions using the paired-end read orientation. Regular paired-end reads are facing inward with a FWD/REV orientation, but when an inversion happens, some reads will be mapping in the opposite orientation regarding the reference. As a consequence, some paired-end reads will have the same orientation: FWD/FWD or REV/REV.
To leverage that information, anvi’o can profile bam files for FWD/FWD and REV/REV reads only with anvi-profile to make special single-profile-db.
anvi-profile -i bam-file \ -c contigs-db \ --fetch-filter inversion
Other essential inputs to run this program
The main input for anvi-report-inversions is a bams-and-profiles-txt, which is a TAB-delimited file composed of at least four columns:
- Sample name,
- contigs-db,
- single-profile-db generated with the inversion fetch filter,
- bam-file.
If you are interested in also characterizing inversion activity statistics across samples, you will also need to add two more columns into the bams-and-profiles-txt file to point out the paths for the R1 and R2 FASTQ files.
Here is a standard run with default parameters:
anvi-report-inversion -P bams-and-profiles-txt \ -o inversions_output
Identifying regions of interest
While anvi’o could directly search for inverted repeats in all the contigs, it would be a waste of time as many IRs are actually not related to inversions. Instead, anvi’o uses the FWD/FWD and REV/REV reads to identify regions of interest and constrain the search for IRs only in these regions.
For this step, you can set the minimum coverage of FWD/FWD and REV/REV reads to define ‘stretches’ with --min-coverage-to-define-stretches. Lower threshold yield more stretches, but also more noise.
The parameter --min-stretch-length defines the minimum length for a stretch to be considered.
FWD/FWD reads are found on the leftmost side of an inversion, while the REV/REV reads are found on the right side. When an inversion is quite long, a region of low to no coverage can separate the two groups of reads. With the flag --min-distance-between-independent-stretches, anvi’o merges fragmented stretches if they are closer than this value.
When the coverage is quite low, a stretch can be a little bit too short and miss potential IRs, so you use --num-nts-to-pad-a-stretch to extend the stretch by x bp upstream and downstream.
Here are the default values for these flags:
anvi-report-inversions -P bams-and-profiles-txt \ -o inversions_output \ --min-coverage-to-define-stretches 10 \ --min-stretch-length 50 \ --min-distance-between-independent-stretches 2000 \ --num-nts-to-pad-a-stretch 100
Finding palindromes
There are a few parameters to constrain the search for palindromic sequences of the IRs, like a minimum length that can be set with --min-palindrome-length, and a maximum number of mismatches with --max-num-mismatches.
You can set the minimum distance between two palindromic sequences with --min-distance. A distance of 0 would correspond to an in-place palindrome, though they don’t relate to genomic inversions.
When searching for palindromes with mismatches, the algorithm will extend the palindrome length as much as possible, often including mismatches which are outside of the true palindrome sequences. The flag --min-mismatch-distance-to-first-base allows you to trim the palindrome when one or more mismatches are n nucleotides away from a palindrome’s start or stop. The default value is 1, meaning that a palindrome MMMMMM(X)M, where M denotes matching nucleotides and X a mismatch, will be trimmed to the first 6 matches MMMMMM.
There are currently two algorithms to find palindromes in anvi’o: numba and BLAST. Numba is very fast when looking for palindromes in short sequences, and BLAST is more efficient for longer stretches. Anvi’o dynamically set the algorithm according to each stretch length: numba for stretches under 5,000 bp and BLAST for longer stretches. You can use the flag --palindrome-search-algorithm to ask anvi’o to use either of these methods explicitly. Note that results between the two methods may differ.
anvi-report-inversions -P bams-and-profiles-txt \ -o inversions_output \ --min-palindrome-length 10 \ --max-num-mismatches 1 \ --min-distance 50 \ --min-mismatch-distance-to-first-base 1 \ --palindrome-search-algorithm numba
Confirming inversions
Multiple palindromes are usually reported for each stretch and to confirm which one actually relates to an inversions, anvi’o searches short-reads in the bam file for unique constructs that can only occur when a genomic region inverted.
By default, anvi’o reports the first confirmed palindrome and moves to the next stretch. This process is very efficient as a stretch usually has only one inversion. But in rare cases, you can have multiple inversions happening in a single stretch. Then, you can use the flag --check-all-palindromes and anvi’o will look for inversion evidence in the short-reads for every palindrome in a stretch.
Anvi’o looks for inversion evidence in the FWD/FWD and REV/REV reads first. If no evidence is found, then it searches the rest of the reads mapping to the region of interest. If you want to only search the FWD/FWD and REV/REV reads you can use the flag --process-only-inverted-reads
Computing inversion activity
If you provide the short-reads R1 and R2 in the bams-and-profiles-txt, anvi’o can compute the proportion of the inversion’s orientation in each sample.
This is a very time consuming step, and if you have multiple sample, you can use the parameter --num-threads to set the maximum of threads for multithreading when possible.
To compute the inversion’s ratios, anvi’o designs in silico primers based on the palindrome sequence and the upstream/downstream genomic context to search short-reads in the raw fastq files. The variable --oligo-primer-base-length is used to control how much of the palindrome should be used to design the primers. The longer, the more specific, but if it is too long, fewer reads will match to the primer.
This step is very computationally intense, but you can test it with the parameter --end-primer-search-after-x-hits. Once the total number of reads reach this parameter, anvi’o will stop searching further and will continue with the next sample. This flag is only good for testing.
If you want to skip this step, you can use the flag --skip-compute-inversion-activity.
An example command with 12 threads:
anvi-report-inversions -P bams-and-profiles-txt \ -o inversions_output \ --num-threads 12 \ --oligo-primer-base-length 12
Computing inversion activity using previously computed inversions
It is possible to instruct anvi’o to use previously reported inversions to characterize their activity across a larger set of samples. This is possible by passing the program anvi-report-inversions the output file for consensus inversions (i.e., ‘CONSENSUS-INVERSIONS.txt’) or the output file for sample-specific inversions (i.e., ‘INVERSIONS-IN-[SAMPLE-NAME].txt’) from a previous run using the flag --pre-computed-inversions:
anvi-report-inversions -P bams-and-profiles-txt \ --pre-computed-inversions inversions_output/INVERSIONS-CONSENSUS.txt -o activity_calculations
In this mode, anvi-report-inversions will not recalculate inversions, and only report the activity of inversions found in the input file across samples listed in the bams-and-profiles-txt file.
If you have additional samples to make sense of the inversions you already know about, you obviously don’t need to do any additional mapping and profiling. All you need is the contigs-db with which you used to generate the inversion report you already have (i.e., not for the new samples you have), and the r1 and r2 files for the new samples. This is similar to the structure of bams-and-profiles-txt, except the profile_db_path and bam_file_path columns. While the entire purpose of this file is ot have these columns, in this particular instance, when you use the flag --pre-computed-inversions with anvi-report-inversions, ANVI’O SANITY CHECKS WILL NOT REQUIRE THE FILE TO INCLUDE PROFILE-DB AND BAM FILE PATHS. Which will make your life much more easier to scale your search. Which means for this particular application, you can create your bams-and-profiles-txt file only with the columns and you’d still be OK: name, contigs_db_path, r1, r2.
Reporting genomic context around inversions
For every inversion, anvi’o can report the surrounding genes and their function as additional files.
You can use the flag --num-genes-to-consider-in-context to choose how many genes to consider upstream/downstream of the inversion. By default, anvi’o report three genes downstream, and three genes upstream.
To select a specific gene caller, you can use --gene-caller. The default is pyrodigal-gv.
If you want to skip this step, you can use the flag --skip-recovering-genomic-context.
Searching for conserved motifs in inverted-repeats
The inversions are carried out by site-specific recombinase which recognize a DNA motifs on each end of the invertible DNA fragment. This DNA motif is a relatively conserved palindrome separated by a short sequence called a spacer.
A fascinating aspect of site-specific recombinases is that they can invert multiple site, providing the DNA recognition motif is present. Some genomes can have one site-specific recombinase responsible many inversions! Some genomes have a few site-specific recombinase regulating different set of inversions.
When you use this command, anvi’o will leverage the program MEME to identify palindrome motifs in the inverted-repeats. It is done in two steps:
- Per inversion. Anvi’o searches and report three motifs. You can find the output of MEME in each inversion’s directory (e.g.
PER_INV/INV_0001/MEME). - For all inversions. Anvi’o searches for shared and conserved motifs across all inversions. By default it will search for as many motifs as there are inversions. This can be time consuming if you have a lot of inversions, and you can use the flag
--num-of-motifsto specify the number of motifs to be reported. The output of MEME can be found here:PER_INV/ALL_INVERSIONS/MEME.
In the final outputs, we report the ‘motif group’ for each inversions. Which means that you can link together groups of inversion putatively associated with the same site-specific recombinase.
If you want to skip this step, you can use the flag --skip-search-for-motifs.
Targeted search
If you are interested in a given contig region you can use the following flags to limit the search:
--target-contig: contig of interest,--target-region-start: the start position of the region of interest,--target-region-end: the end position of the region of interest.
HTML output
The first output of interest is index.html which is a static html page with all the information about each inversions.
On the top of this page, you will find general informations such as number of inversions found and link to some tab-delimited files that summarize all inversions:
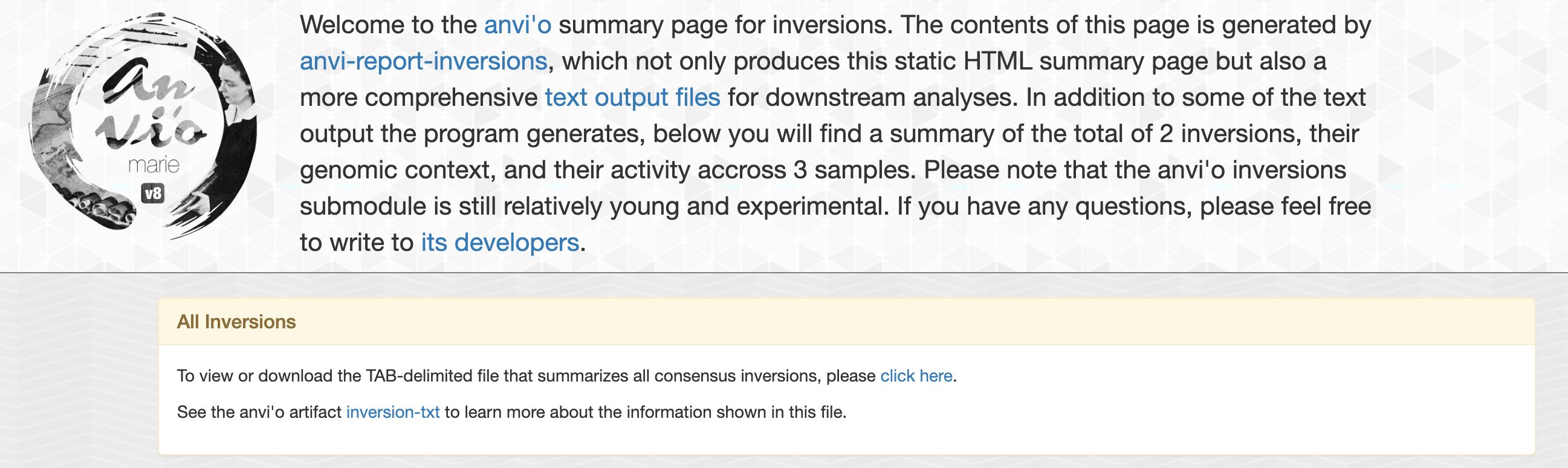
Genomic context
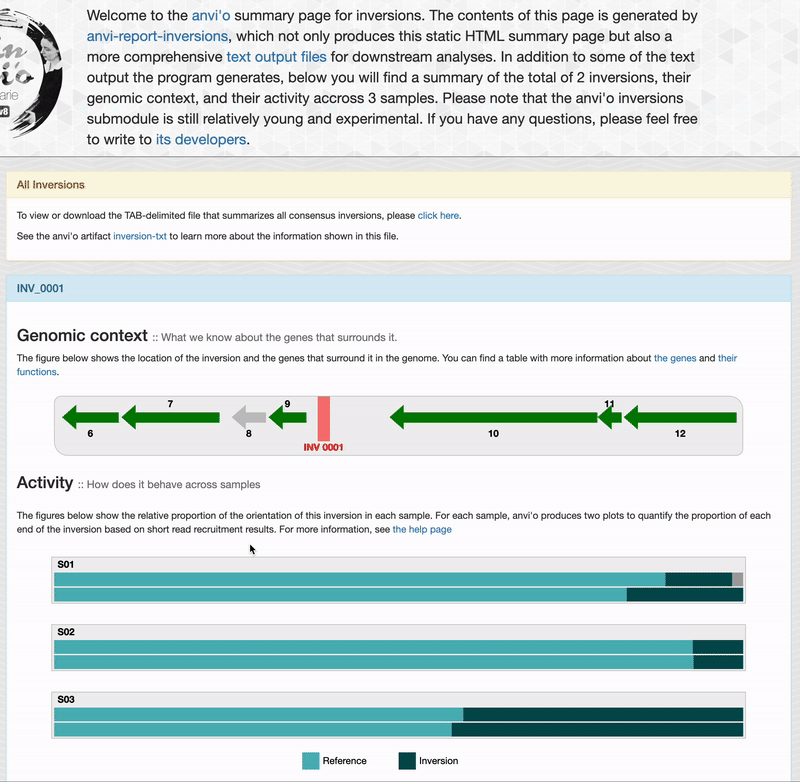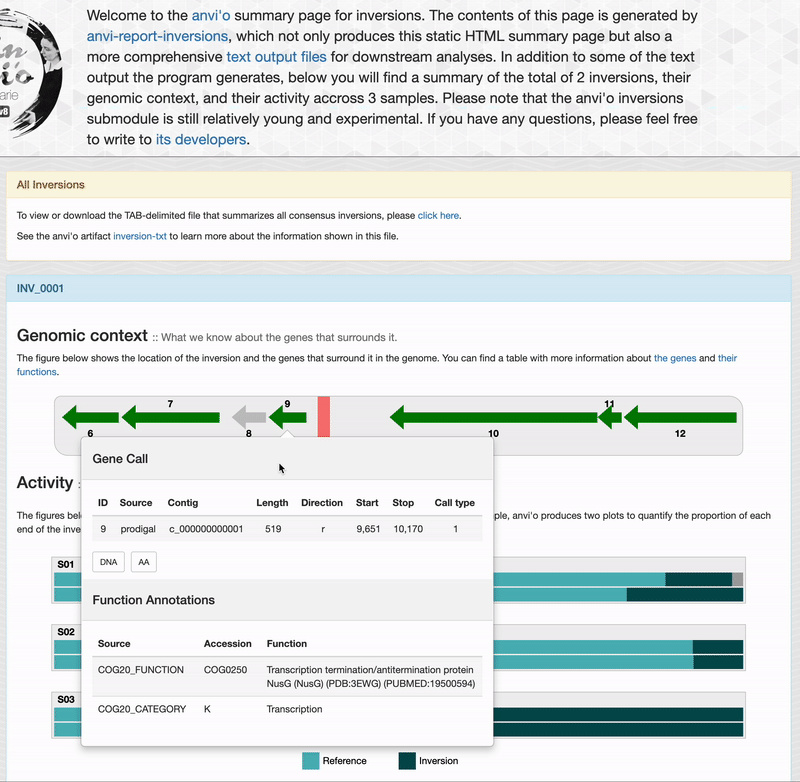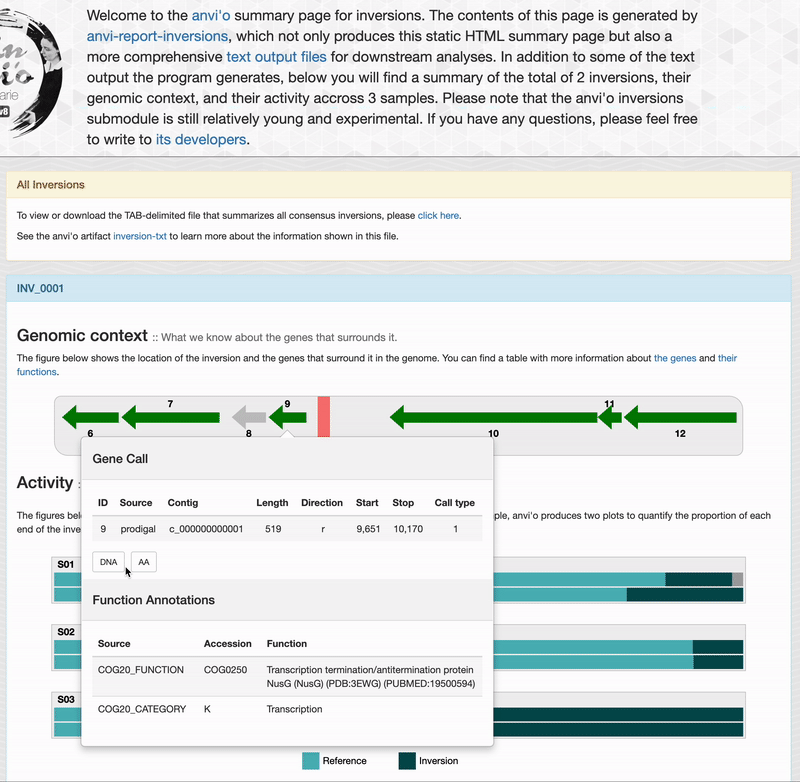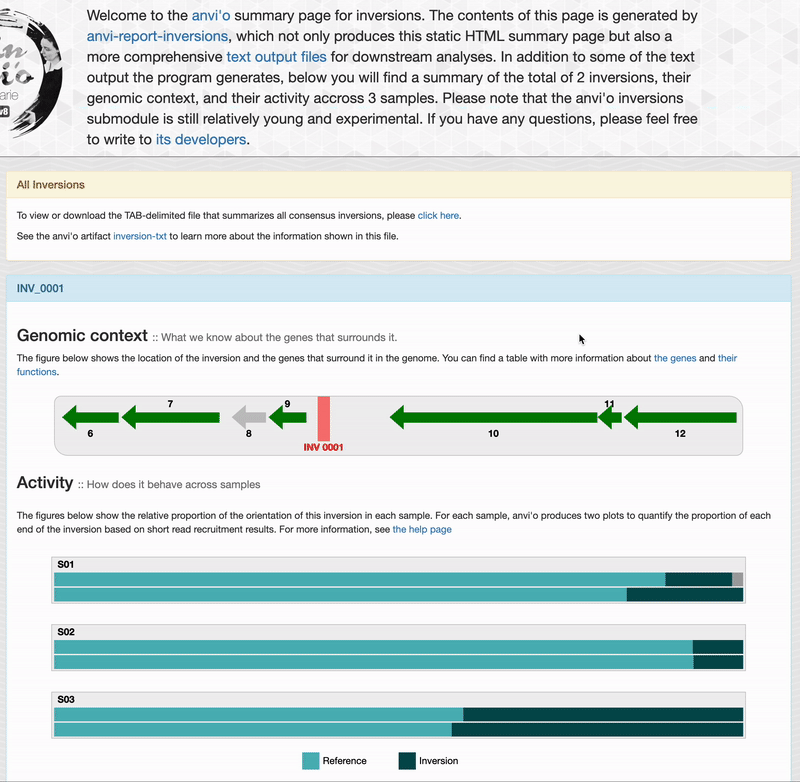
For each inversions you can see the genomic context. This part is interactive and allow you to inspect genes information such as their functions, DNA and amino acid sequences:
Activity
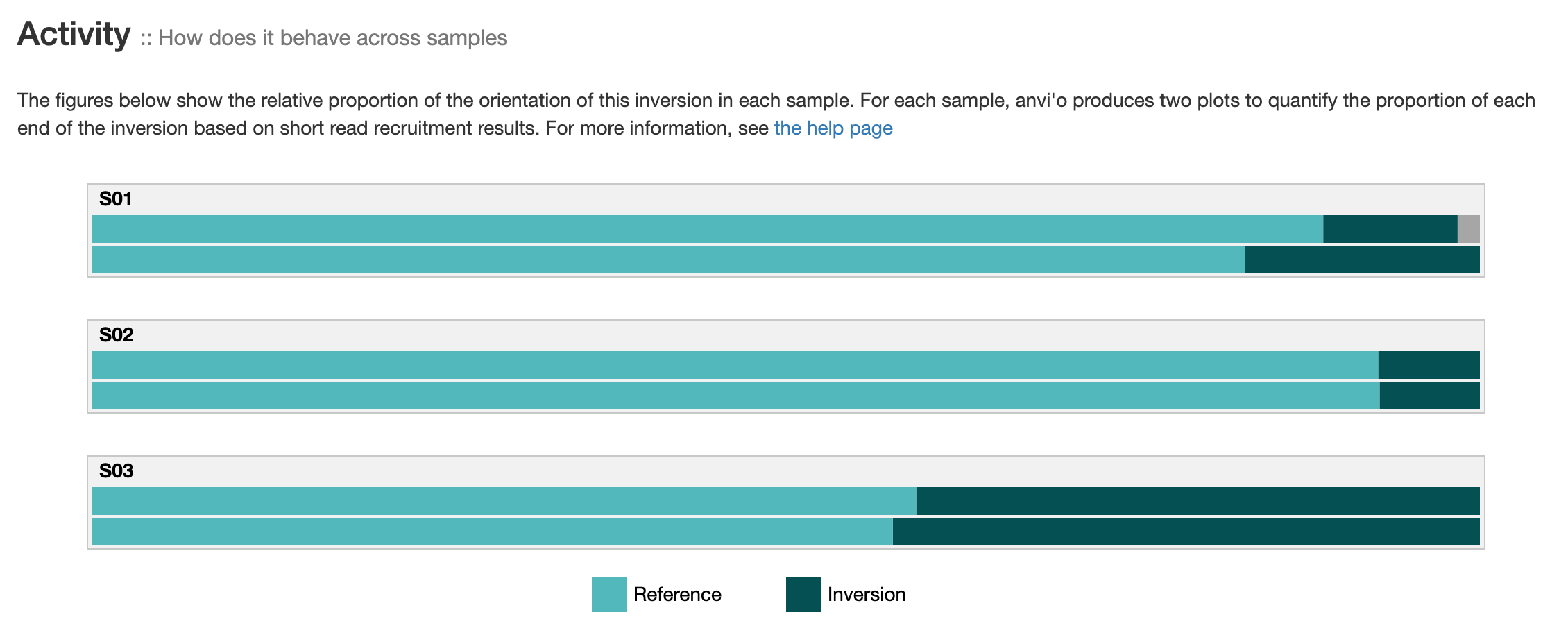
If you provided short-reads to the analysis, you can see barplots representing the inversion’s activity. As explained in the activity section, anvi’o compute the activity on each end of an inversion, which explains the two barplots per sample. “Reference” correspond to the relative proportion of short-read matching to an inversion’s orientation similar to your reference sequence. And “Inversion” correspond to the relative proportion of short-reads matching the reverse orientation.
The two barplots per sample should be relatively similar. It is not the case when (1) the coverage is low (fewer reads = less accurate relative proportions) or (2) there is more than one pair of inverted-repeat in this genomic site. Here is a nice example of multiple nested IRs. If you ever see this, you should consider re-running the command for that specific region with targeted search, and searching for all possible palindrome with --check-all-palindromes. Yes, anvi’o can report nested, multiple IRs inversions when you use these flags :rainbow:.
It is possible to see more colors than the two “Reference” and “Inversion”. For instance, if you have a nested multiple IRs inversions (lucky you), you should see another color (a variation of grey) indicating that more than one sequence can end up on this side of an inversions. Another reason is sequencing error. Anvi’o relies on exact matches to report “Reference” and “Inversion”, and a single read mismatch can create a third category (and therefore a third color in the barplot). If you see some red color, it means more than 4 different DNA sequence can be found at the edge of an inversions, which means that something is probably wrong and requires some manual inspection (or you should collect your Nobel prize).
General information
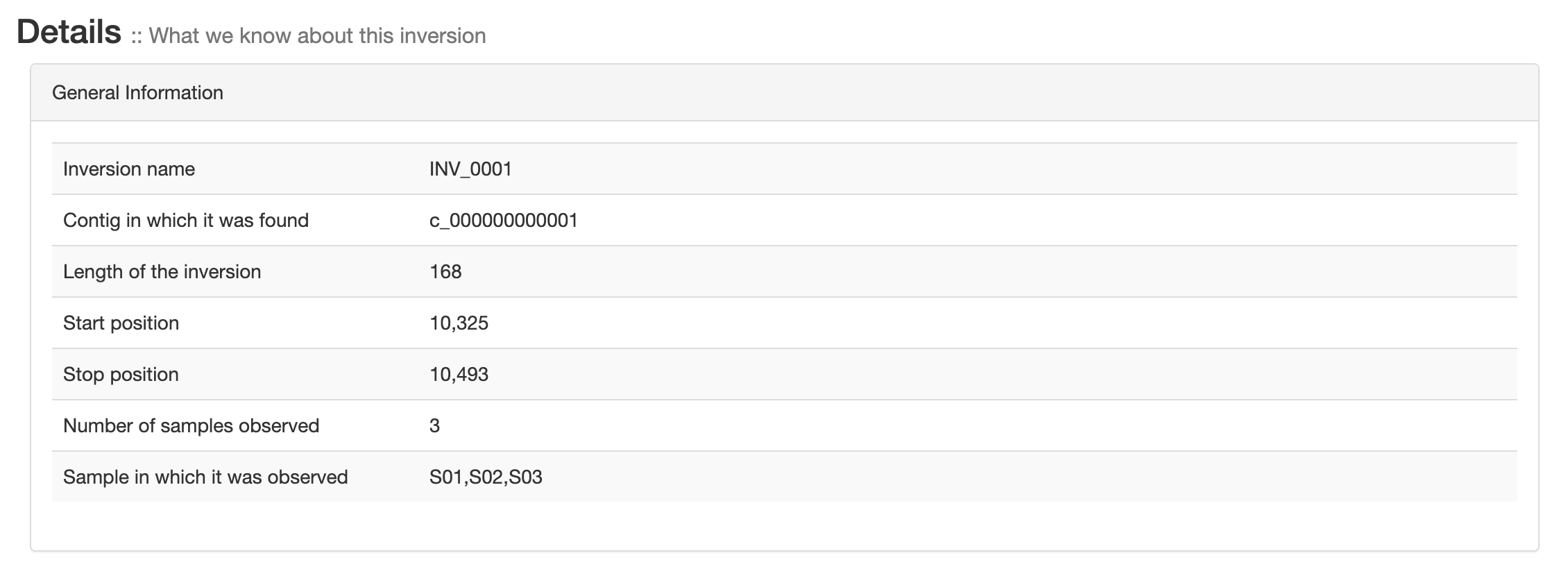
General information about each inversions like contig, position, length and the sample in which it was found.
Inverted repeats and motifs
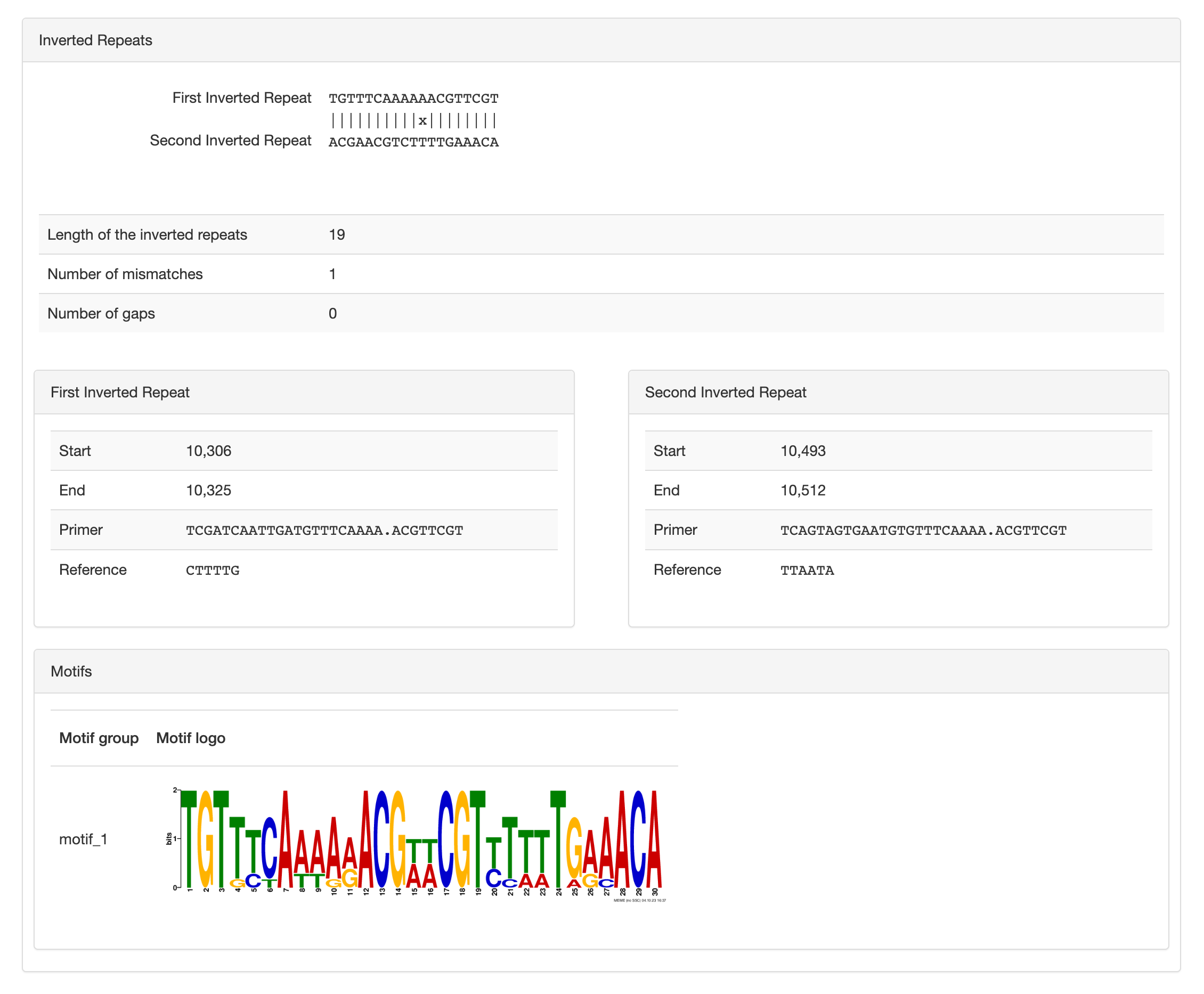
Here you can see the two inverted-repeats, their alignment, length and position. You can also see the “primer” sequence used to search short-read to report the activity, or relative proportion of an inversion’s orientation per sample.
And finally you can see the palindromic motif likely recognized by a site-specific recombinase which carries out such inversion. The same motif can occur in multiple inversions, which suggest that a unique site-specific recombinase is responsible for a set of inversions. Typically there is only one motif per inversion, but we don’t control biology so it is possible that more than one motif are reported.
Other outputs
anvi-report-inversions create an output directory with the following structure (if you have not use the --skip-[..] flags):
INVERSIONS-OUTPUT/
├── ALL-STRETCHES-CONSIDERED.txt
├── INVERSION-ACTIVITY.txt
├── INVERSIONS-CONSENSUS.txt
├── PER_INV
│ ├── ALL_INVERSIONS
│ │ ├── MEME
│ │ │ ├── logo1.eps
│ │ │ ├── [...]
│ │ │ └── meme.xml
│ │ ├── inverted_repeats.fasta
│ │ └── run-MEME.log
│ ├── INV_0001
│ │ ├── MEME
│ │ │ ├── logo1.eps
│ │ │ ├── [...]
│ │ │ └── meme.xml
│ │ ├── SURROUNDING-FUNCTIONS.txt
│ │ ├── SURROUNDING-GENES.txt
│ │ ├── inverted_repeats.fasta
│ │ └── run-MEME.log
│ └── INV_0002
│ ├── MEME
│ │ ├── logo1.eps
│ │ ├── [...]
│ │ └── meme.xml
│ ├── SURROUNDING-FUNCTIONS.txt
│ ├── SURROUNDING-GENES.txt
│ ├── inverted_repeats.fasta
│ └── run-MEME.log
├── PER_SAMPLE
│ ├── INVERSIONS-IN-S01.txt
│ ├── INVERSIONS-IN-S02.txt
│ └── INVERSIONS-IN-S03.txt
└── index.html
For more information about each of these txt files, go check inversions-txt.
Edit this file to update this information.
Additional Resources
Are you aware of resources that may help users better understand the utility of this program? Please feel free to edit this file on GitHub. If you are not sure how to do that, find the __resources__ tag in this file to see an example.