hmm-source
Table of Contents
![]()
A HMM-type anvi’o artifact. This artifact is typically provided by the user for anvi’o to import into its databases, process, and/or use.
🔙 To the main page of anvi’o programs and artifacts.
Provided by
anvi-script-gen-defense-finder-models-to-hmm-directory anvi-script-hmm-to-hmm-directory anvi-script-kofam-accessions-to-hmms-directory anvi-script-pfam-accessions-to-hmms-directory
Required by
anvi-get-sequences-for-hmm-hits
Can be used by
anvi-compute-completeness anvi-delete-hmms anvi-gen-hmm-hits-matrix anvi-run-hmms anvi-script-filter-hmm-hits-table anvi-script-get-hmm-hits-per-gene-call
Description
An HMM source is a collection of one or more hidden Markov models. HMM sources can be used to identify and recover genes in a contigs-db that match to those described in the model.
Models in a given HMM source can be searched in a given contigs-db via the program anvi-run-hmms which would yield an hmm-hits artifact. An anvi’o installation will include multiple HMM sources by default. But HMMs for any set of genes can also be put together by the end user and run on any anvi’o contigs-db.
HMM hits in a contigs-db for a given hmm-source source will be accessible to anvi’o programs globally. Sequences that match to HMM hits can be recovered in an aligned or non-aligned fashion as fasta files for downstream analyses including phylogenomics, they can be displayed in anvi’o interfaces, reported in summary outputs, and so on.
Running anvi-db-info on a contigs-db will list HMM sources available in it.
Default HMM sources
An anvi’o installation will include multiple HMM sources by default. These HMM sources can be run on any contigs-db with anvi-run-hmms to identify and store hmm-hits:
The default HMM sources in anvi’o include:
- Bacteria_71: 71 single-copy core genes for domain bacteria that represent a modified version of the HMM profiles published by Mike Lee. The anvi’o collection excludes
Ribosomal_S20p,PseudoU_synth_1,Exonuc_VII_S,5-FTHF_cyc-lig,YidDandPeptidase_A8occurred in Lee collection (as they were exceptionally redundant or rare among MAGs from various habitats), and includesRibosomal_S3_C,Ribosomal_L5,Ribosomal_L2to make it more compatible with Hug et al’s set of ribosomal proteins. - Archaea_76: 76 single-copy core genes for domain archaea by Mike Lee.
- Protista_83: 83 single-copy core genes for protists (domain eukarya) by Tom O. Delmont.
Apart from these, anvi’o also includes a number of HMM profiles for individual ribosomal RNA classes derived from Torsten Seemann’s tool (we split them into individual classes after this):
- Ribosomal_RNA_5S (eukarya + archaea + bacteria; also includes 5.8S).
- Ribosomal_RNA_12S (mitochondria)
- Ribosomal_RNA_16S (bacteria + archaea + mitochondria)
- Ribosomal_RNA_18S (eukarya)
- Ribosomal_RNA_23S (bacteria + archaea)
- Ribosomal_RNA_28S (eukarya)
When anvi-run-hmms is run on an anvi’o contigs-db without providing any further arguments, it automatically utilizes all the default HMM sources.
Similar to Ribosomal RNAs, anvi’o can also identify Transfer RNAs. Even though Transfer RNAs will also appear as an HMM source for all downstream analyses, their initial identification will require running anvi-scan-trnas program on a contigs-db.
User-defined HMM sources
The user can employ additional HMM sources to identify matching genes in a given contigs-db.
Any directory with expected files in it will serve as an HMM source:
anvi-run-hmms -c contigs-db \ --hmm-source /PATH/TO/USER-HMM-DIRECTORY/
Anvi’o will expect the HMM source directory to contain six files (see this for an example directory). These files are explicitly defined as follows:
- genes.hmm.gz: A gzip of concatenated HMM profiles. One can (1) obtain one or more HMMs by computing them from sequence alignments or by downloading previously computed ones from online resources such as Pfams, (2) concatenate all profiles into a single file called
genes.hmm, and finally (3) compress this file usinggzip. - genes.txt: A TAB-delimited file that must contain three columns:
gene(gene name),accession(gene accession number (can be anything unique)), andhmmsource(source of HMM profiles listed in genes.hmm.gz). The list of gene names in this file must perfectly match to the list of gene names in genes.hmm.gz. - kind.txt: A flat text file which contains a single word identifying what type of profile the directory contains. This information will appear in interfaces. Use a single, descriptive word for your collection.
- reference.txt: A file containing source information for this profile to cite it properly.
- target.txt: A file that specifies the target alphabet and context that defines how HMMs should be searched (this is a function of the HMM source that is used). The proper notation is ‘alphabet:context’. Alphabet can be
AA,DNA, orRNA. Context can beGENEorCONTIG. The content of this file should be any combination of one alphabet and one context term. For instance, if the content of this file isAA:GENE, anvi’o will search genes amino acid sequences, and so on. An exception isAA:CONTIG, which is an improper target since anvi’o can’t translate contigs to amino acid sequences. See this for more details. Please note that HMMs that targetDNA:CONTIGwill result in new gene calls in the contigs database to describe their hits. - noise_cutoff_terms.txt: A file to specify how to deal with noise. See this comment for more information on the contents of this file.
A special note for people who want to create models for use with anvi-run-hmms --add-to-functions-table: Please make sure that each model in your genes.hmm.gz file contains an accession line (starts with ACC) in its header.
Creating anvi’o HMM sources from ad hoc PFAM accessions
It is also possible to generate an anvi’o compatible HMMs directory for a given set of PFAM accession ids. For instance, the following command will result in a new directory that can be used immediately with the program anvi-run-hmms:
anvi-script-pfam-accessions-to-hmms-directory --pfam-accessions-list PF00705 PF00706 \ -o AD_HOC_HMMs
These IDs can be given through the command line as a list, or through an input file where every line is a unique accession id.
An example. Let’s assume we have a genome or a metagenome that looks like this:
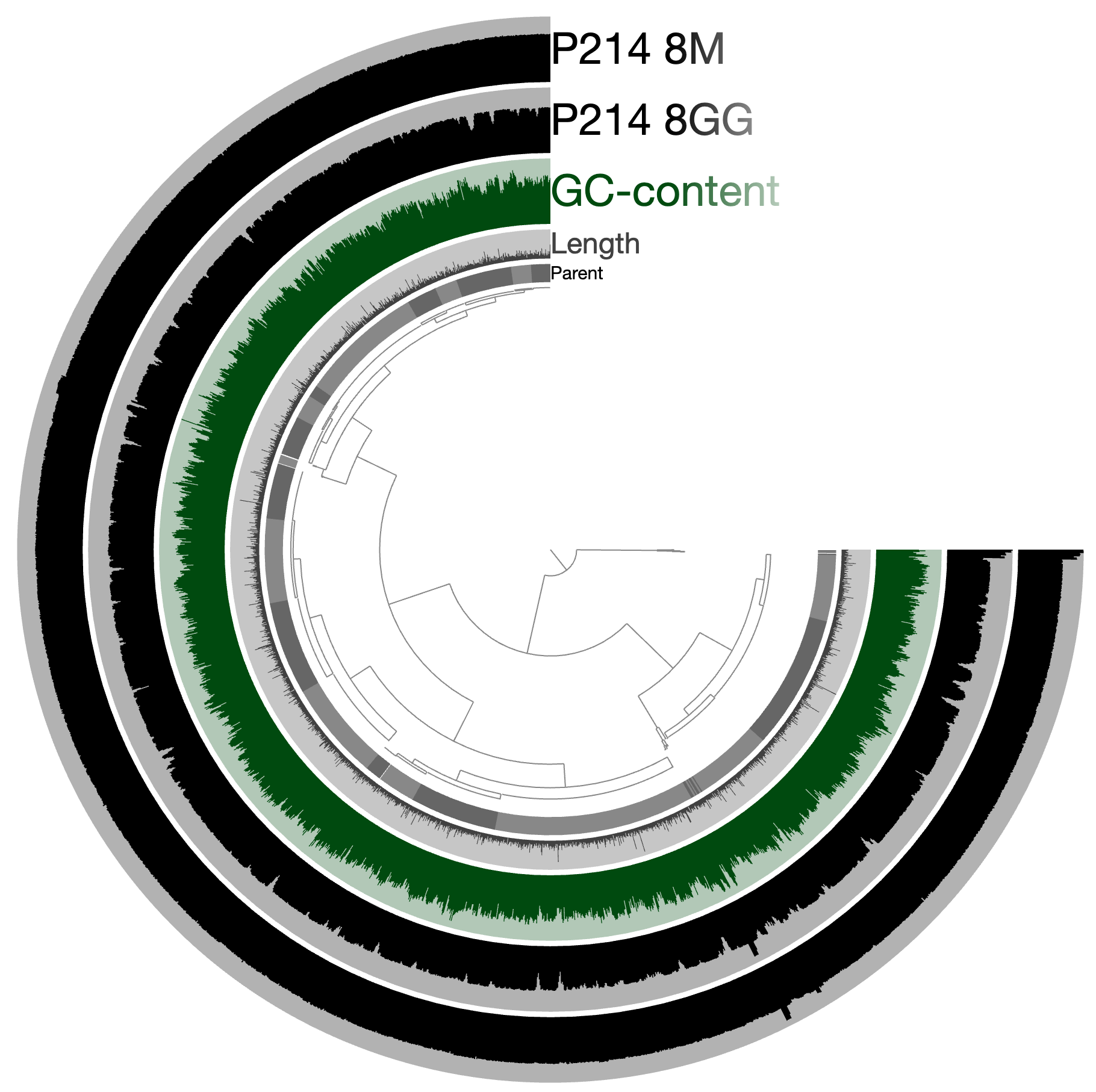
And we wish to identify locations of genes that match to this model: http://pfam.xfam.org/family/PF06603
One can run this command:
anvi-script-pfam-accessions-to-hmms-directory --pfam-accessions-list PF06603 \ -o UpxZ
which would createa a directory called UpxZ. Then, one would run this command to find matches to this model in a given contigs database:
anvi-run-hmms -c CONTIGS.db \ -H UpxZ/ \ --num-threads 4
Now it is possible to get the sequences matching to this model:
anvi-get-sequences-for-hmm-hits -c CONTIGS.db \ --hmm-source UpxZ \ -o UpxZ.fa
Contigs DB ...................................: Initialized: CONTIGS.db (v. 19)
Hits .........................................: 8 hits for 1 source(s)
Mode .........................................: DNA sequences
Genes are concatenated .......................: False
Output .......................................: UpxZ.fa
Or see where they are by visualizing the project using again:
anvi-interactive -p PROFILE.db \ -c CONTIGS.db
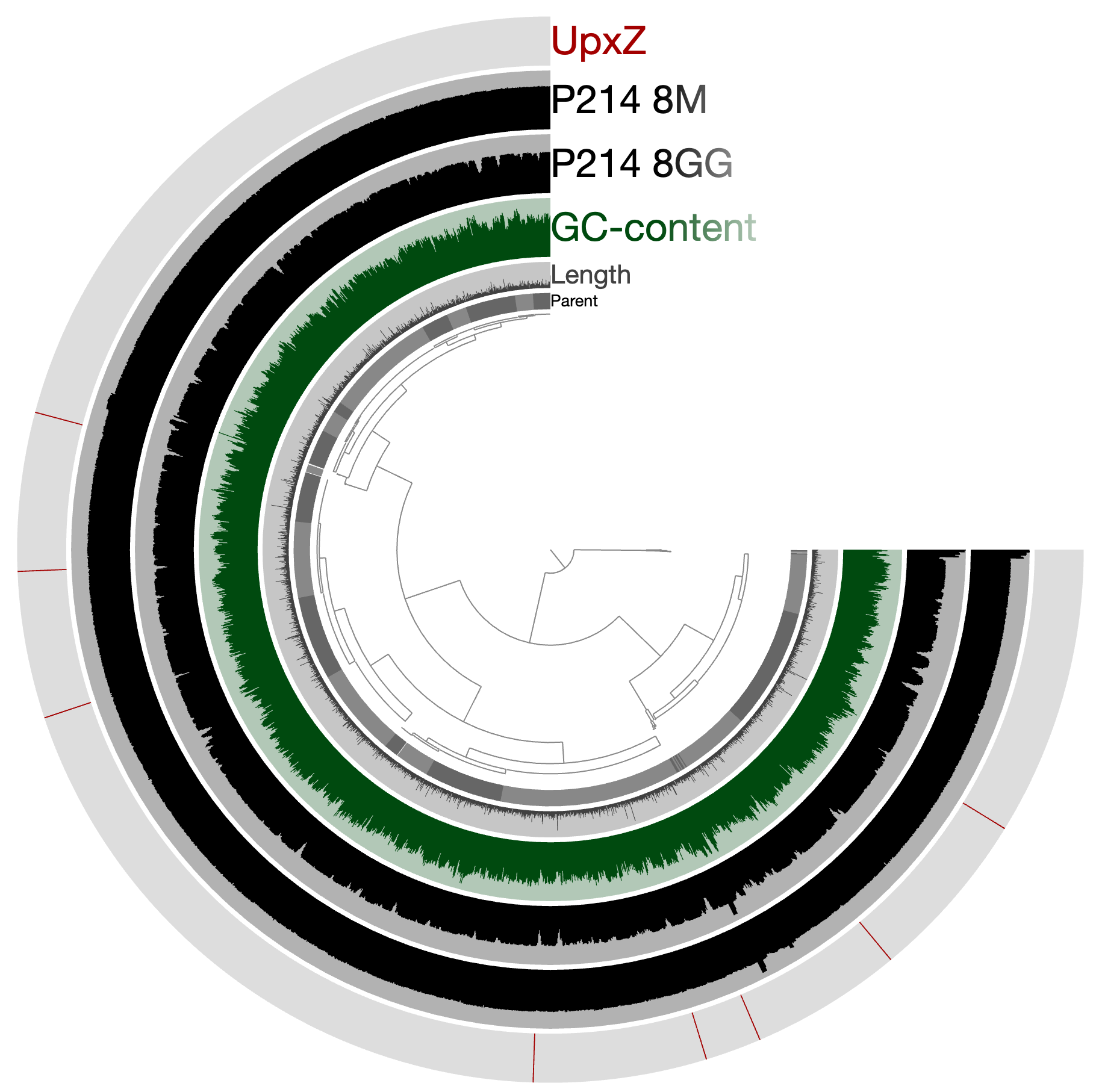
Edit this file to update this information.