The anvi'o 'metagenomics' workflow
Table of Contents
- Authors
- Artifacts accepted
- Artifacts produced
- Third party programs
- Workflow description and usage
- Default mode
- All against all mode
- References Mode
- Long-read and mixed read-type support
- Providing third-party tools via conda
- Running binning algorithms
- Generating summary and split profiles
- Reference-based short read removal
- Frequently Asked Questions
- Is it possible to just do QC and then stop?
- Can I skip anvi-script-reformat-fasta?
- How can I use an existing contigs-db in references mode?
- What’s going on behind the scenes before we run IDBA-UD?
- Can I change the parameters of samtools view?
- Can I change the parameters for Bowtie2?
- How can I restart a failed job?
- Can I use results from previous runs of krakenuniq?
- How do I skip the QC of the FASTQ files?
- Can I use BAM files as input for the metagenomics workflow?
- What to do when submitting jobs with a SLURM system
- How to use metaSPAdes for assembly
From FASTA and/or FASTQ files to anvi'o contigs and profile databases
This workflow is for anyone who wants to do some high-throughput read recruitment. It has a few different modes depending on your input data. By default, it works with metagenomic short reads and will assemble those, use the assembly as a reference for read-recruitment, and create databases ready for anvi-interactive (i.e. for binning). If you don't need to assemble anything, you can also just give this program a set of reference contigs for read-recruitment. Regardless, you'll end up with at least one contigs database + merged profile database pair. Oh, and since this workflow internally uses the contigs workflow, you can optionally run annotation programs on your contigs database as well.
🔙 To the main page of anvi’o programs and artifacts.
Authors


Artifacts accepted
The metagenomics can typically be initiated with the following artifacts:
workflow-config ![]() samples-txt
samples-txt ![]() fasta-txt
fasta-txt ![]()
Artifacts produced
The metagenomics typically produce the following anvi’o artifacts:
Third party programs
This is a list of programs that may be used by the metagenomics workflow depending on the user settings in the workflow-config ![]() :
:
- illumina-utils (Quality control of short reads)
- IDBA-UD (Assembly)
- metaSPAdes (Assembly)
- MEGAHIT (Assembly)
- samtools (BAM file manipulations)
- pyrodigal-gv (Gene calling)
- HMMER (HMM search)
- krakenuniq (Gene taxonomy)
- centrifuge (Gene taxonomy)
- Bowtie2 (Read recruitment)
An anvi’o installation that follows the recommendations on the installation page will include all these programs. But please consider your settings, and cite these additional tools from your methods sections.
Workflow description and usage
The default entering point to the metagenomics workflow is the raw sequencing reads for one or more shotgun metagenomes. The default end point of the workflow is an anvi’o merged profile database ready for refinement of bins (or whatever it is that you want to do with it), along with an annotated anvi’o contigs database. While these are the default entry and end points, there are many more ways to use the metagenomics workflow that we will demonstrate later.
The workflow includes the following steps (though many are optional and can be skipped by modifying their rules in the workflow configuration file):
-
Quality control of metagenomic short reads using illumina-utils, and generating a comprehensive final report for the results of this step (so you have your Supplementary Table 1 ready).
-
Taxonomic profiling of short reads using krakenuniq. These profiles are also imported into individual profile databases, and are available in the merged profile database (for more details about this, refer to the release notes of anvi’o version 5.1).
-
Individual or combined assembly of quality filtered metagenomic reads using either megahit, metaspades, idba_ud or (meta)Flye for long-reads.
-
Generating an anvi’o contigs database from assembled contigs using anvi-gen-contigs-database. This part of the metagenomics workflow is inherited from the contigs workflow, so you know this step also includes the annotation of your contigs database(s) with functions, HMMs, and taxonomy.
-
Mapping short reads from each metagenome to the contigs using bowtie2, or long-reads with minimap2, and generating sorted and indexed BAM files.
-
Profiling individual BAM files using anvi-profile to generate single anvi’o profiles.
-
Merging resulting single anvi’o profiles using anvi-merge.
The metagenomics workflow is quite talented and can be run in multiple ‘modes’. The following sections will detail different use cases.
This documentation page is partially based off of this old tutorial. We hope to update it at some point, but this is what we have for now. You will notice that several sections below reference example files and data. If you want to run those examples yourself, you should click here and follow the instructions to download the mock dataset used in that tutorial.
Default mode
As mentioned above, the standard usage of this workflow is meant to go through all the steps from raw reads to having a merged profile database (or databases) ready for binning.
All you need is a bunch of FASTQ files, and a samples-txt file. Here, we will go through a mock example with three small metagenomes. These metagenomes were made by choosing a small number of reads from three HMP metagenomes (these reads were not chosen randomly, for more details, ask Alon). In your working directory you have the following samples.txt file:
$ column -t samples.txt
sample group r1 r2
sample_01 G01 three_samples_example/sample-01-R1.fastq.gz three_samples_example/sample-01-R2.fastq.gz
sample_02 G02 three_samples_example/sample-02-R1.fastq.gz three_samples_example/sample-02-R2.fastq.gz
sample_03 G02 three_samples_example/sample-03-R1.fastq.gz three_samples_example/sample-03-R2.fastq.gz
This is the file that describes our ‘groups’ and locations of raw paired-end reads for each sample. The default name for your samples-txt file is samples.txt, but you can use a different name by specifying it in the config file (see below).
In your working directory there is a config file called config-idba_ud.json; let’s take a look at it.
{
"workflow_name": "metagenomics",
"config_version": "2",
"samples_txt": "samples.txt",
"anvi_script_reformat_fasta": {
"run": true,
"--prefix": "{group}",
"--simplify-names": true,
"--keep-ids": "",
"--exclude-ids": "",
"--min-len": "",
"--seq-type": "",
"threads": ""
},
"idba_ud": {
"--min_contig": 1000,
"threads": 11,
"run": true
}
}
Relatively short. Every configurable parameter (and there are many many of them) that is not mentioned here will be assigned a default value.
We usually like to start with a default config file, and edit parameters that are important to us. Usually these edits are related to making true values false if we don’t want to run a particular step, or change number of threads assigned to a single step, etc.
So what do we have in the example config file above?
-
samples_txt: Path for our
samples.txt(since we used the default namesamples.txt, we didn’t really have to include this in the config file, but it is always better to be explicit). -
idba_ud: A few parameters for
idba_ud.-
run: Currently two assembly software packages are available in the workflow: megahit and idba_ud. We didn’t set either of these as the default program, and hence if you wish to assemble things then you must set the
runparameter totruefor one (and only one) of these. -
–min-contig: From the help menu of
idba_udwe learn thatidab_udhas the default as200, and we want it as1,000, and hence we include this in the config. -
threads: When you wish to use multi-threads you can specify how many threads to use for each step of the workflow using this parameter. Here we chose 11 threads for
idba_ud.
-
{kind=link}
A note on rule-specific parameters We suggest that you take a minute to look at the default config file. To do so, run:
anvi-run-workflow -w metagenomics \
--get-default-config default-metagenomics-config.json
It is very long, and that’s why we didn’t paste it here. We keep things flexible for you, and that means having many parameters.
But there are some general things you can notice:
-
threads - every rule has the parameter “threads” available to it. This is meant for the case in which you are using multi-threads to run things. To learn more about how snakemake utilizes threads you can refer to the snakemake documentation. We decided to allow the user to set the number of threads for all rules, including ones for which we ourselves never use more than 1 (why? because, why not? maybe someone would one day need it for some reason. Don’t judge). When threads is the only parameter that is available for a rule, it means that there is nothing else that you can configure for this rule. Specifically, it means you don’t even get to choose whether this rule is run or not. But don’t worry, snakemake will make sure that steps that are not necessary will not run.
-
run - some rules have this parameter. The rules that have this parameter are optional rules. To make sure that an optional rule is run you need to set the
runparameter totrue. If you wish not to run an optional rule, then you must setruntofalseor simply an empty string (""). Some of the optional rules run by default and others don’t. You can find out what the default behavior is by looking at the default config file. As mentioned above, if a rule doesn’t have the run parameter it means that snakemake will infer whether it needs to run or not (just have some trust please!). -
parameters with an empty value (
"") - Many of the parameters in the default config file get an empty value. This means that the default parameter that is provided by the underlying program will be used. For example, the ruleanvi_gen_contigs_databaseis responsible for running anvi-gen-contigs-database (we tried giving intuitive names for rules :-)). Below you can see all the available configurations foranvi_gen_contigs_database. Let’s take the parameter--split-lengthas an example. By refering to the help menu of anvi-gen-contigs-database you will find that the default for--split-lengthis 20,000, and this default value will be used by anvi-gen-contigs-database if nothing was supplied in the config file. You may notice another interesting thing, which is that the value for--project-nameis"{group}". This is a little magic trick to make it so that the project name in your contigs database would be indentical to the group name that you supplied in the config file. If you wish to understand this syntax, you may read about the snakemake wildcards.
(...)
"anvi_gen_contigs_database": {
"--project-name": "{group}",
"threads": 5,
"--description": "",
"--skip-gene-calling": "",
"--ignore-internal-stop-codons": "",
"--skip-mindful-splitting": "",
"--contigs-fasta": "",
"--split-length": "",
"--kmer-size": ""
},
(...)
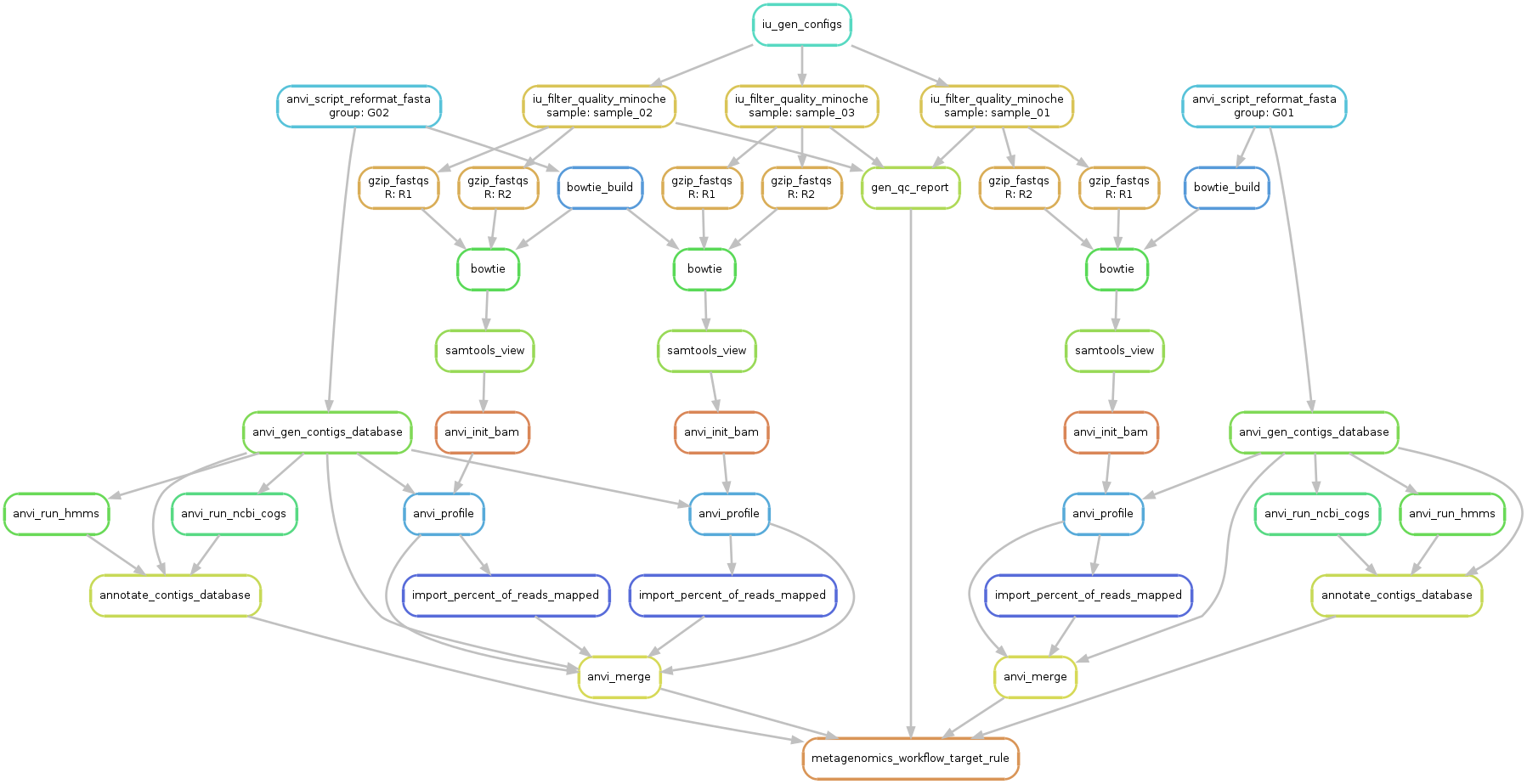
Ok, so now we have everything we need to start. Let’s first run a sanity check and create a workflow graph for our workflow:
anvi-run-workflow -w metagenomics \
-c config-idba_ud.json \
--save-workflow-graph
A file named workflow.png was created and should look like this:
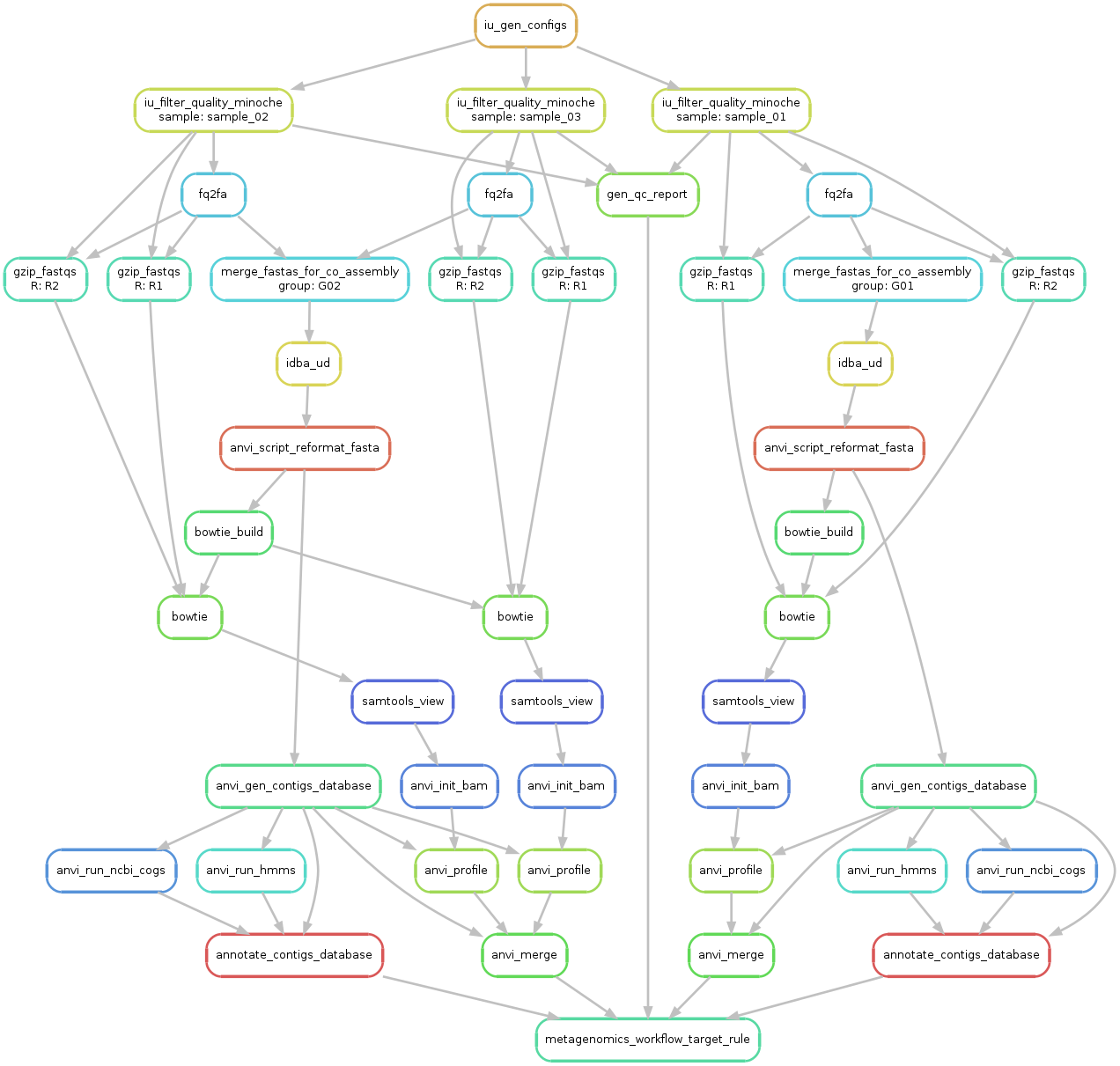
Take a minute to take a look at this image to understand what is going on. From a first look it might seem complicated, but it is fairly straightforward (and also, shouldn’t you know what is going on with your data?!?).
Ok, let’s run this.
Using “screen” to run stuff
We always start our work by initiating a screen session. If you are not familiar with what this is, basically, we use it here because we are running something that requires no user interaction for a long time on a remote machine (e.g. a cluster head node).
screen -S mysnakemakeworkflow
After the workflow is running you simply click ctrl-A followd by D to detach from the screen. If you want to check the status of your workflow, then to reconnect to your screen use:
screen -r mysnakemakeworkflow
And when you want to kill it use CTRL-D (while connected to the screen).
At any given time you can see a list of all your screens this way:
screen -ls
Simple, but extremely efficient.
Now we can run the workflow:
anvi-run-workflow -w metagenomics \
-c config-idba_ud.json
Once everything finishes running (on our cluster it only takes 6 minutes as these are very small mock metagenomes), we can take a look at one of the merged profile databases:
anvi-interactive -p 06_MERGED/G02/PROFILE.db \
-c 03_CONTIGS/G02.db
And it should look like this:
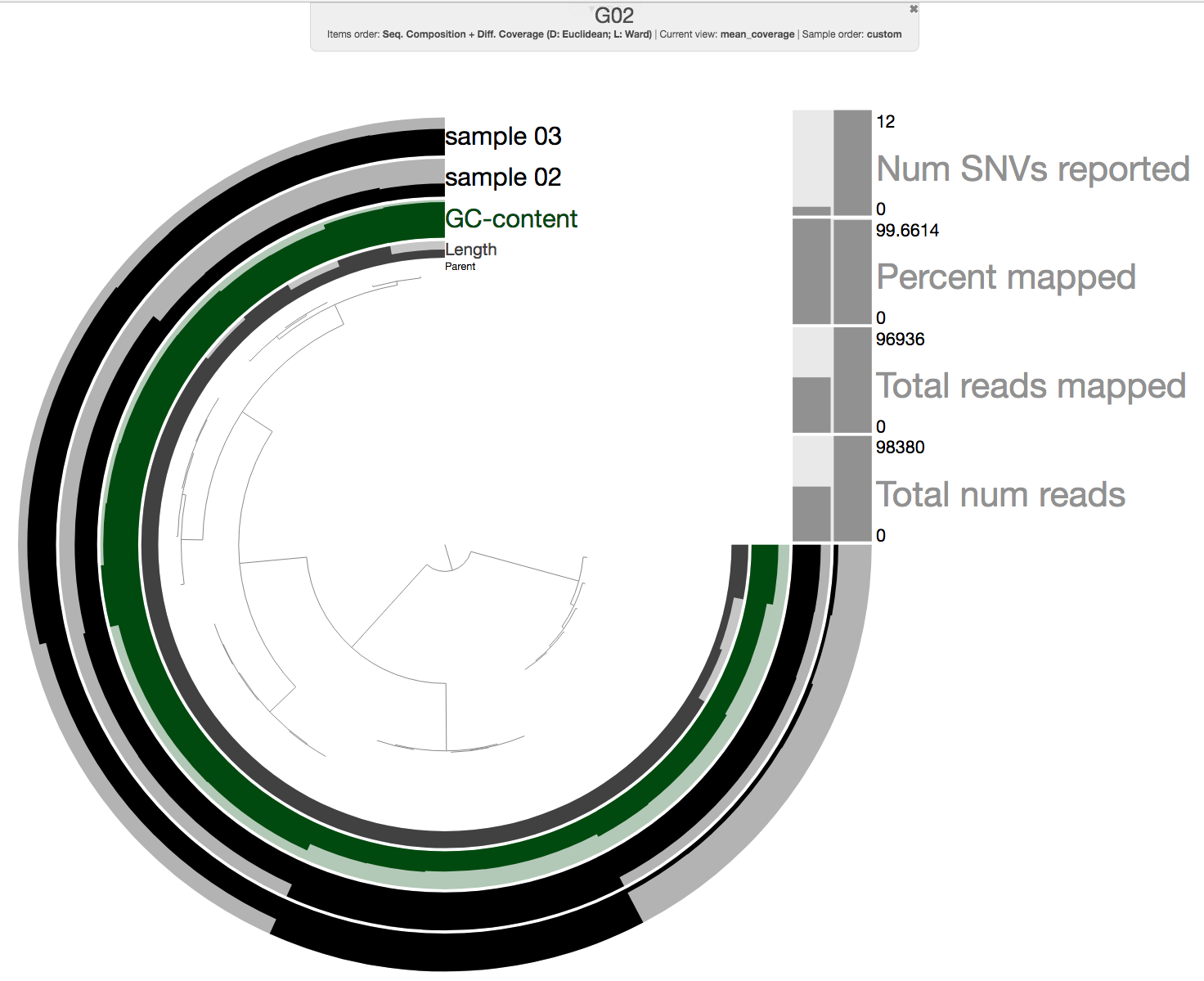
Ok, so this looks like a standard merged profile database with two samples. As a bonus, we also added a step to import the number of reads in each sample (“Total num reads”), and we also used it to calculate the percentage of reads from the sample that have been mapped to the contigs (“Percent Mapped”).
This is a bit of an expert knowledge, but if you remember, we had two “groups” in the samples.txt file. Hence, we have two contigs databases for G01 and G02. But one of our groups had only a single sample, there was nothing to merge. Thus, there is no merged profile for G01 at the location you would expect to find it, but instead, there is a README file there:
$ cat 06_MERGED/G01/README.txt
Only one file was profiled with G01 so there is nothing to
merge. But don't worry, you can still use anvi-interactive with
the single profile database that is here: 05_ANVIO_PROFILE/G01/sample_01/PROFILE.db
Which means, while you can use the program anvi-interactive to interactively visualize merged profile databases that are affiliated with groups that have more than one sample, you will find profiles to visualize under single profiles directories for groups associated with a single sample (such as G01 in our example):
anvi-interactive -p 05_ANVIO_PROFILE/G01/sample_01/PROFILE.db \
-c 03_CONTIGS/G01.db
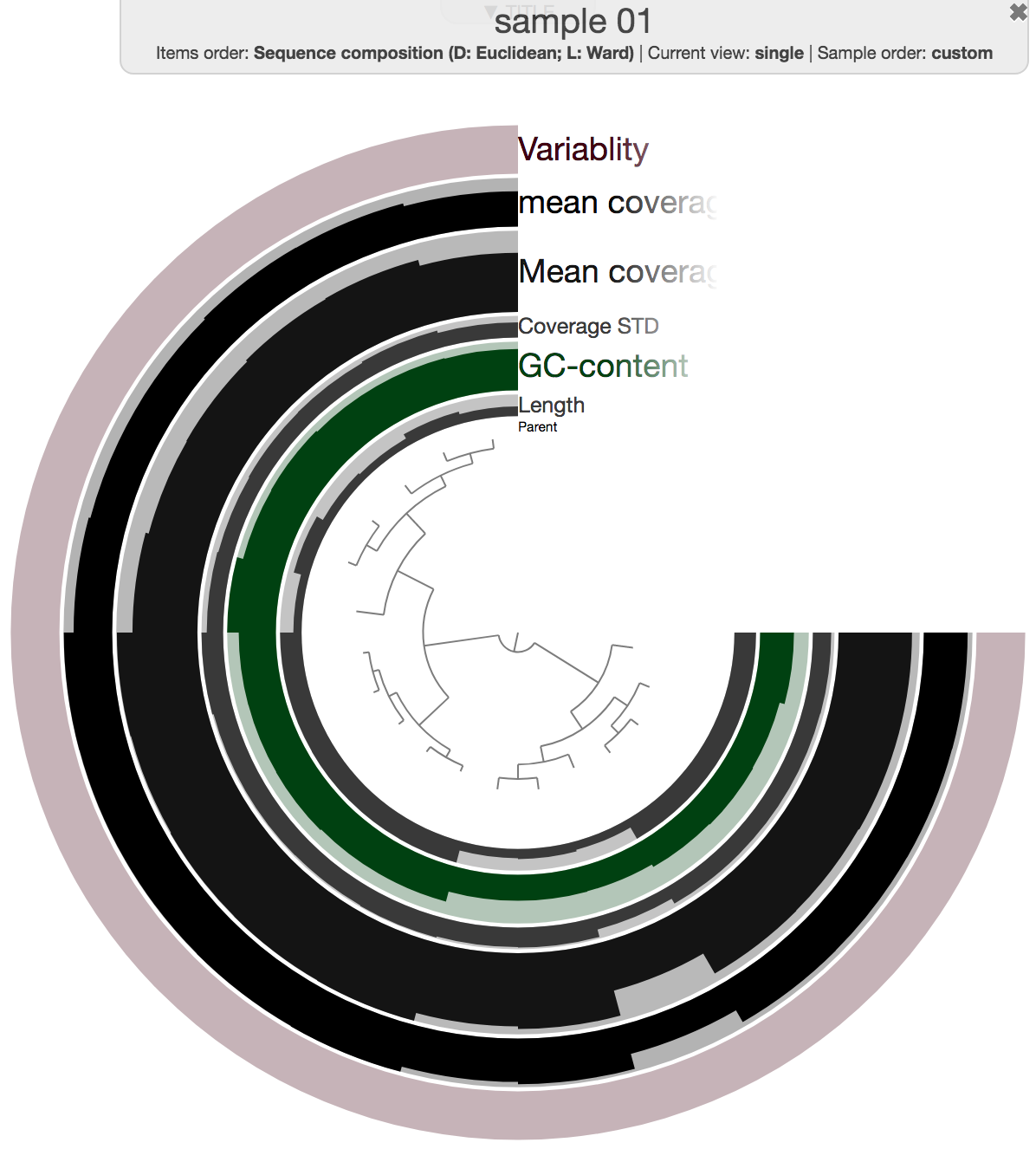
A note on directory structure
The default directory structure that will appear in the working directory includes these directories:
00_LOGS
01_QC
02_FASTA
03_CONTIGS
04_MAPPING
05_ANVIO_PROFILE
06_MERGED
The 00_LOGS directory contains a subdirectory for the workflow run, such as 00_LOGS/metagenomics or 00_LOGS/idba_ud if the config names the run through LOGS_DIR. Within that directory, logs are organized by workflow rule, such as 00_LOGS/metagenomics/anvi_profile/ or 00_LOGS/metagenomics/anvi_merge/. Each rule-specific directory contains the log files for individual jobs. The workflow also writes 00_LOGS/metagenomics/metagenomics-workflow-manifest.tsv, a tab-delimited file that records whether each job succeeded or failed and points to the relevant rule log.
Don’t like these names? You can specify the name of the folder by providing the following information in the config file:
(...)
"output_dirs": {
"LOGS_DIR" : "00_MY_beAuTiFul_LOGS",
"QC_DIR" : "BEST_QC_DIR_EVER",
"ASSEMBLY_DIR": "assemblies",
"CONTIGS_DIR" : "/absolute/path/to/my/contigs/dir",
"MAPPING_DIR" : "relative/path/to/my/mapping/dir",
"PROFILE_DIR" : "/I/already/did/my/profiling/and/this/is/where/you/can/find/it/",
"MERGE_DIR". : "06_Keep_Calm_and_Merge_On"
}
(...)
You can change all, or just some of the names of these output folders. And you can provide an absolute or a relative path for them.
In addition to the merged profile databases and the contigs databases (and all intermediate files), the workflow has another output, the QC report, which you can find here: 01_QC/qc-report.txt. Let’s look at it:
| sample | number of pairs analyzed | total pairs passed | total pairs passed (percent of all pairs) | total pair_1 trimmed | total pair_1 trimmed (percent of all passed pairs) | total pair_2 trimmed | total pair_2 trimmed (percent of all passed pairs) | total pairs failed | total pairs failed (percent of all pairs) | pairs failed due to pair_1 | pairs failed due to pair_1 (percent of all failed pairs) | pairs failed due to pair_2 | pairs failed due to pair_2 (percent of all failed pairs) | pairs failed due to both | pairs failed due to both (percent of all failed pairs) | FAILED_REASON_P | FAILED_REASON_P (percent of all failed pairs) | FAILED_REASON_N | FAILED_REASON_N (percent of all failed pairs) | FAILED_REASON_C33 | FAILED_REASON_C33 (percent of all failed pairs) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sample_01 | 10450 | 8423 | 80.6 | 0 | 0 | 0 | 0 | 2027 | 19.4 | 982 | 48.45 | 913 | 45.04 | 132 | 6.51 | 0 | 0 | 2027 | 100 | 0 | 0 |
| sample_02 | 31350 | 25550 | 81.5 | 0 | 0 | 0 | 0 | 5800 | 18.5 | 2777 | 47.88 | 2709 | 46.71 | 314 | 5.41 | 0 | 0 | 5800 | 100 | 0 | 0 |
| sample_03 | 60420 | 49190 | 81.41 | 0 | 0 | 0 | 0 | 11230 | 18.59 | 5300 | 47.2 | 5134 | 45.72 | 796 | 7.09 | 0 | 0 | 11230 | 100 | 0 | 0 |
All against all mode
The default behavior for this workflow is to create a contigs database for each group and map (and profile, and merge) the samples that belong to that group. If you wish to map all samples to all contigs, use the all_against_all option in the config file:
"all_against_all": true
In your working directory you can find an updated config file config-idba_ud-all-against-all.json, which looks like this:
{
"workflow_name": "metagenomics",
"config_version": 1,
"samples_txt": "samples.txt",
"idba_ud": {
"--min_contig": 1000,
"threads": 11,
"run": true
},
"all_against_all": true
}
And we can generate a new workflow graph:
anvi-run-workflow -w metagenomics \
-c config-idba_ud-all-against-all.json \
--save-workflow-graph
An updated DAG for the workflow for our mock data is available below:
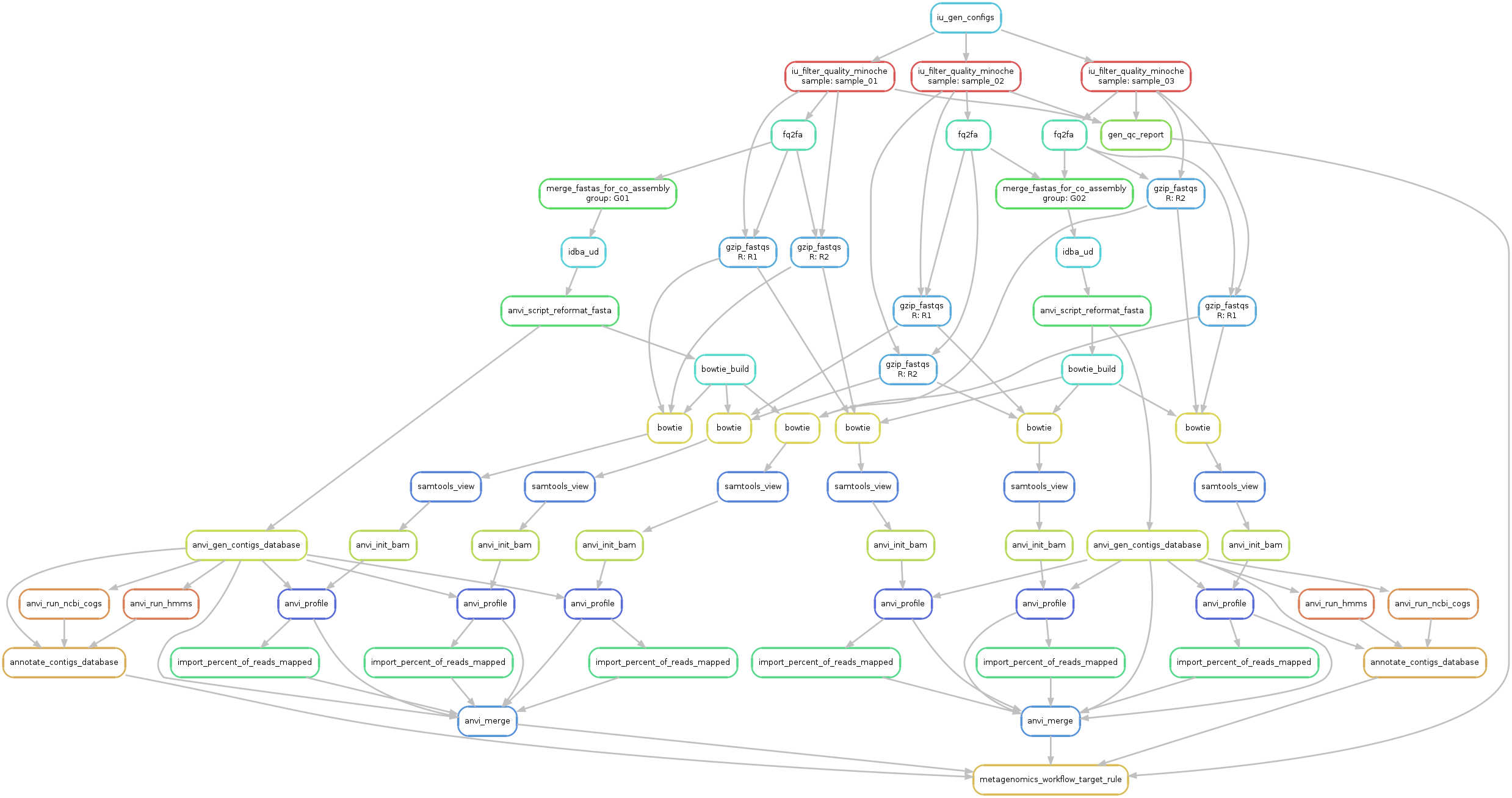
A little more of a mess! But also has a beauty to it :-).
A short advertisement for snakemake If you are new to snakemake, you might be surprised to see how easy it is to switch between modes. All we need to do is tell the anvi_merge rule that we want all samples merged for each group, and snakemake immediately infers that it needs to also run the extra mapping, and profiling steps. Thank you snakemake! (says everyone).
References Mode
This mode is used when you have one or more genomes, and one or more metagenomes from which you wish to recruit reads using your genomes.
Along with assembly-based metagenomics, we often use anvi’o to explore the occurrence of population genomes across metagenomes. A good example of how useful this approach could be is described in this blogpost: DWH O. desum v2: Most abundant Oceanospirillaceae population in the Deepwater Horizon Oil Plume.
For this mode, what you have is a bunch of FASTQ files (metagenomes) and FASTA files (reference genomes), and all you need to do is to let the workflow know where to find these files, using two .txt files: samples_txt, and fasta_txt.
fasta_txt should be a 2 column tab-separated file, where the first column specifies a reference name and the second column specifies the file path of the FASTA file for that reference.
After properly formatting your samples_txt and fasta_txt, reference mode is initiated by adding these to your config file:
(...)
"references_mode": true
(...)
The samples_txt stays as before, but this time the group column will specify for each sample, which reference should be used (aka the name of the reference as defined in the first column of fasta_txt). If the samples_txt file doesn’t have a group column, then an “all against all” mode would be provoked.
In your directory you can find the following fasta.txt, and config-references-mode.json:
$ cat fasta.txt
name path
G01 three_samples_example/G01-contigs.fa
G02 three_samples_example/G02-contigs.fa
$ cat config-references-mode.json
{
"workflow_name": "metagenomics",
"config_version": "2",
"fasta_txt": "fasta.txt",
"samples_txt": "samples.txt",
"references_mode": true,
"output_dirs": {
"FASTA_DIR": "02_FASTA_references_mode",
"CONTIGS_DIR": "03_CONTIGS_references_mode",
"QC_DIR": "01_QC_references_mode",
"MAPPING_DIR": "04_MAPPING_references_mode",
"PROFILE_DIR": "05_ANVIO_PROFILE_references_mode",
"MERGE_DIR": "06_MERGED_references_mode",
"LOGS_DIR": "00_LOGS_references_mode"
}
}
Let’s create a workflow graph:
A note from Alon on why we need the references_mode flag This is a note that is mainly directed at anvi’o developers, so feel free to skip this note.
We could have just invoked “references_mode” if the user supplied a fasta_txt, but I decided to have a specific flag for it, to make things more verbose for the user.
Now we can run this workflow:
anvi-run-workflow -w metagenomics \
-c config-references-mode.json
Long-read and mixed read-type support
The metagenomics workflow handles long reads (Oxford Nanopore, PacBio) alongside — or instead of — short reads. To include long reads, add an lr column to your samples-txt pointing to the long-read FASTQ files (long reads must be FASTQ, not FASTA — the long-read QC and mapping steps rely on per-base quality scores). A single sample can carry both short reads (r1/r2) and long reads (lr); the workflow tracks the two read sets separately and never mixes incompatible read types in assembly or mapping.
Long reads are assembled with (meta)Flye and mapped with minimap2, while short reads continue to use your chosen short-read assembler and bowtie2.
Choosing long-read presets: the lr_technology column vs. the config
Each long-read tool needs a technology-appropriate preset: a read-type flag for Flye (e.g. --nano-raw) and a mapping preset for minimap2 (e.g. map-ont). There are two ways to provide these, and anvi’o will never run these tools on their built-in defaults: if it cannot determine a preset, it stops before building the workflow with a message telling you exactly what to set.
-
Recommended — the
lr_technologycolumn in your samples-txt. If you declare the sequencing technology per sample (e.g.ont,pb-hifi; see the samples-txt documentation for the full list), anvi’o automatically selects the correct preset for every long-read tool. When the column is present it is all-or-nothing: every long-read sample must have a value, and short-read-only samples leave it blank. Samples in the same co-assemblygroupmust use technologies that map to the same Flye read type, otherwise anvi’o asks you to split them into separate groups. -
The config file. If you omit the
lr_technologycolumn, you must set the presets explicitly in your workflow-config:minimap2’spreset, and exactly one of Flye’s read-type flags (--nano-raw,--pacbio-hifi, …).
The technology → preset mapping is maintained in anvio/workflows/lr_technology_presets.yaml, which also records the tool versions each preset was validated against; if you have an untested version installed, anvi’o prints a non-fatal heads-up but proceeds.
Long-read quality control
Beyond the default short-read QC (illumina-utils), the workflow offers several optional QC steps, all disabled by default and enabled per rule in your workflow-config with "run": true:
filtlong— length/quality filtering of long reads with Filtlong (--min-length,--max-length,--target-bases,--keep-percent,--min-mean-q).--min-length/--max-lengthare hard length thresholds, while--target-bases,--keep-percent, and--min-mean-qfilter against the read set’s own length/quality distribution. When enabled, downstream mapping and assembly use the filtered reads. Because Filtlong is a filter, anvi’o requires at least one filtering criterion when it is enabled — set one of the parameters above, or pass another Filtlong option viaadditional_params; otherwise anvi’o stops with an error.nanoplot— long-read quality assessment with NanoPlot (one report per long-read readset per stage). It needs no sequencing-technology preset. Two independent flags control which reads it assesses:run_on_raw(the original reads) andrun_on_filtered(thefiltlongoutput). Both default tofalse, so when you enablenanoplotyou must set at least one of them totrue; set both to get a before/after comparison of your filtering.fastqc— FastQC on short reads (one report per short-read readset per stage). Likenanoplot, it takesrun_on_raw(the original reads) andrun_on_filtered(the illumina-utilsQUALITY_PASSEDreads), both defaulting tofalse.multiqc— aggregates the FastQC and NanoPlot (NanoStats) outputs into a single MultiQC report. Samples are labelled<readset> | <stage>(e.g.S1 | raw,S1 | filtered) so that when both stages are produced, a sample’s raw and filtered rows sort next to each other rather than being split into separateraw/filteredblocks.
For nanoplot and fastqc, anvi’o validates the stage flags before running: if the tool is enabled you must select at least one stage, and run_on_filtered is only allowed when the matching filter (filtlong for nanoplot, iu_filter_quality_minoche for fastqc) is enabled — otherwise there would be no filtered reads to look at. In practice, if you enable one of these tools without any filtering, set run_on_raw: true.
filtlong, nanoplot, fastqc, and multiqc are not shipped inside the anvi’o environment. See Providing third-party tools via conda below for how the workflow gets hold of them (this applies to the assemblers and mappers too).
Providing third-party tools via conda
Several rules in the metagenomics workflow call programs that anvi’o does not bundle in its own environment — the assemblers, mappers, and QC tools:
bowtie (Bowtie2) · minimap2 · megahit · metaspades · idba_ud · flye · filtlong · nanoplot · fastqc (FastQC) · multiqc
By default, anvi’o expects each enabled tool on your $PATH: it checks that the program exists before starting and stops with a clear error if it does not. Alternatively, so you do not have to install every one of these into a single environment, you can have any rule run inside its own conda environment that anvi’o (through Snakemake) sets up just for that rule. You control this per rule in your workflow-config with three mutually exclusive options, all off by default:
-
use_anvio_conda_yaml(boolean; defaultfalse). Set it totrueto use the environment file anvi’o ships for this rule (one curated, version-pinned.yamlper tool, living inanvio/workflows/conda_envs/). The file is resolved at run time from your installed anvi’o, so your config stays reproducible on any machine — there is no hard-coded path to a repository. This is the easiest way to get a pinned, tested version of a tool without installing anything yourself. -
conda_yaml— a path to your own environment.yamlfile (for example, a copy of one of anvi’o’s that you have customized, or an entirely different recipe). Snakemake builds the environment from it. -
conda_env— the name of an environment you have already created yourself (e.g.conda create -n my-flye -c bioconda -c conda-forge flye). The rule runs the tool viaconda run -n <name> ....
When a rule resolves to a conda .yaml (the anvi’o-shipped one via use_anvio_conda_yaml: true, or your own conda_yaml), anvi’o automatically adds --use-conda to the underlying Snakemake command, so Snakemake builds and activates that environment. The first run builds the environment (which takes a little while); later runs reuse the cached environment. Because conda_env points at an environment that already exists, it does not need --use-conda.
These three options are mutually exclusive
Set at most one of use_anvio_conda_yaml: true, conda_yaml, or conda_env per rule; if more than one is in effect anvi’o stops with a clear error rather than guessing which one you meant. For example, to run Flye from your own named environment:
"flye": {
"run": true,
"conda_env": "my-flye"
}
Running these tools from your $PATH
This is the default. With all three conda options off for a rule (use_anvio_conda_yaml: false and both conda_yaml and conda_env empty), anvi’o expects the program on your $PATH and checks for it up front.
Note that whenever a rule is set to be provided by conda (any of the three options above), anvi’o will not pre-check that the program exists on your $PATH — Snakemake will provide it. In that case, if the environment cannot be built or the tool is otherwise unavailable, you will see the failure when that rule runs rather than up front.
Running binning algorithms
If you wish to utilize automatic binning algorithms, you can use anvi-cluster-contigs as part of your metagenomics workflow. You can run one or more binning algorithms, and resulting collections would be automatically imported into your merged profile database/s.
The configuration parameters for the anvi_cluster_contigs rule look like this by default:
(...)
"anvi_cluster_contigs": {
"run": "",
"--driver": "",
"--collection-name": "{driver}",
"--just-do-it": "",
"--additional-params-concoct": "",
"--additional-params-metabat2": "",
"--additional-params-maxbin2": "",
"--additional-params-dastool": "",
"--additional-params-binsanity": "",
"threads": ""
},
(...)
Let’s go over how to work with these:
- run - you must set this to
true(no quotation marks) if you wish to run this rule. - –driver - you can choose one or more from the list of binning algorithms that are available with anvi-cluster-contigs. To see what is available run
anvi-cluster-contigs -h. If you wish to use multiple algorithms you must provide a list with the proper format. For example:[ "concoct", "metabat2" ](notice that each algorithm name is inside quotation, but the brackets are not). - –collection-name - You can see that by default, this is set to
"{driver}". We recommend just leaving it as-is. Using the curly brackets like this is a special way to let Snakemake know that this is a “wildcard” (basically a keyword). If you are not familiar with Snakemake, no worries. What happens here is that the keyword “driver” is swaped for the algorithm name. So if we chose to run CONCOCT and MetaBAT2, then the names for the collections, by default, would be “concoct” and “metabat2”, respectively. if you wish to change it, you have to include"{driver}"inside your new name (so for example,"{driver}_collection2"is Ok), otherwise, all the algorithms you run will have the same collection name, which means they will try to override each other. If you are using only a single binning algorithm, then feel free to change to collection name to whatever you want (since you don’t need to worry about multiple algorithms overriding each other). - –just-do-it - instructs anvi-cluster-contigs to just run and not bother you with questions and complaints (as much as possible). For example, this would allow anvi-cluster-contigs to override a collection if there was a collection with identical name already in your profile database.
- –additional-params-concoct - this parameter (as well as all the other
--additional-params-parameters) are here so that you can set parameters that are specific to each clustering algorithm. To see which parameters are available refer to the help menu:anvi-cluster-contigs -h. For example, for concoct, we can provide something like this:"--additional-params-concoct": "--clusters 100 --iterations 200".
Generating summary and split profiles
As of anvi’o v5.3, you can also configure the workflow to import collections and generate a summary and/or split your profile database (using anvi-split). If you are running the workflow and plan to do binning, then you would usually not have a collection yet. But often we already have a collection ready (e.g. if you are re-profiling things for some reason, or if you are performing mapping and profiling on a FASTA file that was generated by merging a bunch of genomes into one fasta).
In order for the workflow to import a collection into a merged profile database you need to provide a collection-txt file in the following manner:
"collections_txt": "path/to/YOUR_COLLECTIONS_TXT_FILE"
This is the format for the collections.txt file:
| name | collection_name | collection_file | bins_info | contigs_mode | default_collection |
|---|---|---|---|---|---|
| G01 | MOCK | MOCK-collection.txt | MOCK-collection-info.txt | ||
| G02 | 1 |
Where:
- name: is the name of the group to which the collection corresponds (this should match the names of groups in your
samples_txt(if you supplied these), or the names of references in yourfasta_txt(in references mode). In default mode (AKA assembly mode), if you didn’t supply group names, then the group names are identical to the sample names in yoursamples_txt - The four following columns (
collection_name,collection_file,bins_info,contigs_mode) correspond to parameters of anvi-import-collection. Onlycollection_name, andcollection_fileare mandatory, and the rest of the columns are optional. collection_name: the name for the collection - you must provide a value.collection_file: a path to your collection file (i.e. the file that specifies the bin for each split/contig).bins_info: (optional) a path to your bins-info txt filecontigs_mode: (optional) if your collection file include contigs names (instead of splits) set this column to1.- The last column (
default_collection) is an optional column to specify if you want a default collection to be imported using anvi-script-add-default-collection. If you want the default collection, then set the value in this column to1. The default collection will be calledDEFAULTand the bin name would be the name in thenamecolumn of thecollections.txtfile (i.e. the “group” name).
If you specify you want a default_collection for a group then you can’t specify a collection file for this group (these options are mutually exclusive). In addition, anvi_split will not run for a group with a default collection (a default collection includes a single bin with all the contigs, so there is nothing to split).
Your collections_txt could include only some of your groups, and then collections would be imported only to the merged profile databases that correspond to these group names.
anvi_summarize and/or anvi_split (whichever you configured to run) will run for each group that is specified in your collections.txt.
Let’s run a mock example. We can update the config file for references mode in the following manner to run these steps:
{
"workflow_name": "metagenomics",
"config_version": 1,
"fasta_txt": "fasta.txt",
"references_mode": true,
"collections_txt": "collections.txt",
"anvi_summarize": {
"run": true
},
"anvi_split": {
"run": true
},
"output_dirs": {
"FASTA_DIR": "02_FASTA_references_mode",
"CONTIGS_DIR": "03_CONTIGS_references_mode",
"QC_DIR": "01_QC_references_mode",
"MAPPING_DIR": "04_MAPPING_references_mode",
"PROFILE_DIR": "05_ANVIO_PROFILE_references_mode",
"MERGE_DIR": "06_MERGED_references_mode",
"SUMMARY_DIR": "07_SUMMARY_references_mode",
"SPLIT_PROFILES_DIR": "08_SPLIT_PROFILES_references_mode",
"LOGS_DIR": "00_LOGS_references_mode"
}
}
And we have the following collections.txt:
name collection_name collection_file bins_info contigs_mode
G02 MOCK MOCK-collection.txt MOCK-collection-info.txt
Once we run this, we can find the summary in the following directory: 08_SUMMARY/G02-SUMMARY/.
And for each bin in MOCK-collection.txt we have a directory under: 09_SPLIT_PROFILES/G02/.
Reference-based short read removal
As of anvi’o v5.3, we added a feature for removing short reads based on mapping to reference FASTA files.
The purpose of this feature is to allow you to filter reads that match certain reference genomes. As you will see below, you can also use this feature to just quantify the reads that match these reference FASTA, without removing these reads from the FASTQ files (see dont_remove_just_map).
This step is performed by the rule remove_short_reads_based_on_references. By default, this rule will not run.
Here are the default parameters for this rule:
(...)
"remove_short_reads_based_on_references": {
"delimiter-for-iu-remove-ids-from-fastq": " ",
"dont_remove_just_map": "",
"references_for_removal_txt": "",
"threads": ""
},
(...)
Let’s go over the parameters of this rule:
references_for_removal_txt - This is a table similar to the fasta.txt file, with two columns: reference and path. This rule is performed if and only if a table text file was supplied using this parameter.
dont_remove_just_map - If you set this parameter to true, then the mapping will be performed in order to count the number of reads in each sample that matched the references in your references_for_removal_txt, but that’s it (i.e. these reads will not be removed from your FASTQ files). The reason we decided to add this feature is to let you assess the number of reads that probably match these references, without risking losing reads that actually matter to you. More specifically, this way the assembly step has access to all the reads that were in the FASTQ file. You can see the note by Brian Bushnell here for an example as to why you wouldn’t want to remove short reads (in the method we use for removing them) before your assembly.
delimiter-for-iu-remove-ids-from-fastq - this allows you to set the --delimiter for iu-remove-ids-from-fastq, which is the program we use for the removal of short reads. Refer to the manual (by running iu-remove-ids-from-fastq -h) to better understand this feature. By default we set the --delimiter to a single space " " (we found it to be useful sometimes and harmless in other cases).
The bam files that are created during mapping are saved in MAPPING_DIR/REF_NAME, where REF_NAME is the name you gave to the particular reference in the references_for_removal_txt file.
In your working directory you can find the file mock_ref_for_removal.txt, which looks like this:
reference path
R1 mock_ref_for_removal1.fa
R2 mock_ref_for_removal2.fa
We can modify the config from the References Mode section above (but notice that this mode could be used in the default mode as well).
{
"workflow_name": "metagenomics",
"config_version": 2,
"fasta_txt": "fasta.txt",
"references_mode": true,
"remove_short_reads_based_on_references": {
"delimiter-for-iu-remove-ids-from-fastq": " ",
"dont_remove_just_map": "",
"references_for_removal_txt": "mock_ref_for_removal.txt"
},
"output_dirs": {
"FASTA_DIR": "02_FASTA_references_mode",
"CONTIGS_DIR": "03_CONTIGS_references_mode",
"QC_DIR": "01_QC_references_mode",
"MAPPING_DIR": "04_MAPPING_references_mode",
"PROFILE_DIR": "05_ANVIO_PROFILE_references_mode",
"MERGE_DIR": "06_MERGED_references_mode",
"LOGS_DIR": "00_LOGS_references_mode"
}
}
Now you can run this:
anvi-run-workflow -w metagenomics \
-c config-references-mode-with-short-read-removal.json
Removing Human DNA from a metagenome A common use-case for the reference based short read removal is to filter human DNA from a metagenome.
You can download the human genome from here:
wget ftp://ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/annotation/GRCh38_latest/refseq_identifiers/GRCh38_latest_genomic.fna.gz
And then use the following references_for_removal_txt:
reference path
HUMAN GRCh38_latest_genomic.fna.gz
Frequently Asked Questions
If you need something, send your question to us and we will do our best to add the solution down below.
Is it possible to just do QC and then stop?
If you only want to QC your files and then compress them (and not do anything else), simply invoke the workflow with the following command:
anvi-run-workflow -w metagenomics \
-c config.json \
--additional-params \
--until gzip_fastqs
Can I skip anvi-script-reformat-fasta?
Yes! In “reference mode”, you may choose to skip this step, and keep your original contigs names by changing the anvi_script_reformat_fasta rule the following way:
"anvi_script_reformat_fasta": {
"run": false
}
In assembly mode, this rule is always executed.
How can I use an existing contigs-db in references mode?
This is relevant if you already have a contigs-db, and all you want to do is to recruit reads from a bunch of metagenomes.
This is done through ‘references mode’, but as you see in the relevant section, this mode asks yo to provide a fasta-txt, from which it generates contigs-db files for your references. What if you have your own contigs database, and you do not want to generate a new one? You can achieve that and make snakemake skip the creation of a new contigs database by putting the existing one at the place it is expected to be created. This is an example directory structure you should aim for before starting the workflow:
├── 03_CONTIGS
│ ├── anvi_run_hmms-EXAMPLE.done
│ ├── anvi_run_ncbi_cogs-EXAMPLE.done
│ ├── anvi_run_scg_taxonomy-EXAMPLE.done
│ ├── EXAMPLE-annotate_contigs_database.done
│ └── EXAMPLE.db
├── config.json
├── contigs.fa
├── fasta.txt
└── samples.txt
where,
03_CONTIGSis the directory name defined in your config.json file to store contigs databases (03_CONTIGSis already the default directory name, so name it as such if you didn’t change anything in the config.json).- The
.donefiles in03_CONTIGSinstrcuts anvi’o to not re-run those jobs on the existing contigs databse. Add them withtouchor remove as necessary. config.jsonis yor configuration where you have at least the following entries:
(...)
"fasta_txt": "fasta.txt",
"samples_txt": "samples.txt",
"references_mode": true,
(...)
-
contigs.fais the output of anvi-export-contigs run on your contigs-db in03_CONTIGS. -
fasta.txtis your fasta-txt that contains a single entry with nameEXAMPLEand should look exactly like this:name path EXAMPLE contigs.fa Please note: when you change
EXAMPLEto something more meaningful, you will have to replaceEXAMPLEwith the same name in every other file in the list above. -
samples.txtis your good old samples-txt that contains your metagenomes with which the read recruitment will be conducted.
EASY PEASY.
What’s going on behind the scenes before we run IDBA-UD?
A note regarding idba_ud is that it requires a single FASTA as an input. Because of that, what we do is use fq2fa to merge the pair of reads of each sample to one FASTA, and then we use cat to concatenate multiple samples for a co-assembly. The FASTA file is created as a temporary file, and is deleted once idba_ud finishes running. If this is annoying to you, then feel free to contact us or just hack it yourself. We tried to minimize memory usage by deleting each individual FASTA file after it was concatenated to the merged FASTA file (see this issue for details).
Can I change the parameters of samtools view?
The samtools command executed is:
samtools view additional_params -bS INPUT -o OUTPUT
Where additional_params refers to any parameters of samtools view that you choose to use (excluding -bS or -o, which are always set by the workflow). For example, you could set it to be -f 2, or -f 2 -q 1 (for a full list see the samtools documentation). The default value for additional_params is -F 4.
Can I change the parameters for Bowtie2?
Similar to samtools we use the additional_params to configure Bowtie2. The bowtie rule executes the following command:
bowtie2 --threads NUM_THREADS \
-x PREFIX_OF_BOWTIE_BUILD_OUTPUT \
-1 R1.FASTQ \
-2 R2.FASTQ \
additional_params \
-S OUTPUT.sam
Hence, you can use additional_params to specify all parameters except --threads, -x, -1, -2, or -S.
For example, if you don’t want gapped alignment (aka the reference does not recruit any reads that contain indels with respect to it), and you don’t want to store unmapped reads in the SAM output file, set additional_params to be --rfg 10000,10000 --no-unal (for a full list of options see the bowtie2 documentation).
How can I restart a failed job?
If your job fails for some reason you can use additional_params with the original command to restart the workflow where it stopped. For example:
anvi-run-workflow -w metagenomics \
-c config-idba_ud.json \
--additional-params \
--keep-going \
--rerun-incomplete
Here using additional_params with the --keep-going and --rerun-incomplete flags will resume the job even if it failed in the middle of a rule, like anvi_profile. Of course, it is always a good idea to figure out why a workflow failed in the first place.
When a workflow fails, then you would need to unlock the working directory before rerunning. This means you would have to run the full command with the --unlock flag once, and then run the command again without the --unlock flag. Please refer to the snakemake documentation for more details regarding how snakemake locks the working directory.
Can I use results from previous runs of krakenuniq?
If you already ran krakenuniq on your metagenomes, then you can use the kraken_txt option in the config file to provide a path to a TAB-delimited file with the paths to the tax file for each of your metagenomic samples. Notice that the kraken_txt file must have the following format (i.e. two columns with the headers “sample” and “path”):
sample path
s01 /path/to/s01-kraken.tax
s02 /path/to/s02-kraken.tax
The sample names must be identical to the sample names that are provided in the samples.txt file, and it should include all the samples in samples.txt.
Once you have such a file, and let’s say you named it kraken.txt, simply add this to your config file:
"kraken_txt": "kraken.txt"
How do I skip the QC of the FASTQ files?
If you already ran quality filtering for your FASTQ files, then just make sure that this is included in your config file:
"iu_filter_quality_minoche": {
"run": false
}
Can I use BAM files as input for the metagenomics workflow?
In short, yes. If you already did mapping, and you have a bunch of bam files, and now you want to run additional steps from the workflow (e.g. generate contigs databases, annotate them, profile the bam files, etc.), then it might not be entirely straightforward, but it is possible (and I wish to extend my thanks to Even Sannes Riiser for troubleshooting this process).
This is what you need to do:
- Make sure you have a samples.txt file. The first column is, as usual, the name of your sample. As for the other two columns
r1, andr2, in your case you should no longer need the FASTQ files, and hence this two column could have any arbitrary word, but you still have to have something there (if you still have access to your FASTQ files, and you want to run something like krakenHLL, then in that case, you should put the path to the FASTQ files, just as in the normal case of asamples.txtfile) - You should tell the workflow to skip QC. If you don’t do this, then the workflow by default would look for your FASTQ files, and QC them, and run everything else, including mapping.
- You should use references mode.
- You need to make sure your bam files have names compatible with what the snakemake workflow expects. The way we expect to find the bam file is this:
MAPPING_DIR/group_name/sample_name.bamWhere
MAPPING_DIRis04_MAPPINGby default but you can set it in the config file.group_nameis the name you gave the reference in yourfasta.txtfile. Andsample_nameis the name you gave the sample in thesamples.txtfile. - You must skip
import_percent_of_reads_mapped. Currently, we use the log files of bowtie2 to find out how many reads were in the (Qc-ied) FASTQ files, but since you already did your mapping elsewhere, we don’t know how to get that information, and hence you must skip this step. This is pretty easy to do manually later on, so no big deal. In order to skipimport_percent_of_reads_mapped, include this in your config file:
"import_percent_of_reads_mapped": {
"run": false
}
What to do when submitting jobs with a SLURM system
If you want to work with any cluster managing software (such as SLURM) you just need to use the --cluster argument of snakemake. Here is what the snakemake help menu tells us:
--cluster CMD, -c CMD
Execute snakemake rules with the given submit command,
e.g. qsub. Snakemake compiles jobs into scripts that
are submitted to the cluster with the given command,
once all input files for a particular job are present.
The submit command can be decorated to make it aware
of certain job properties (input, output, params,
wildcards, log, threads and dependencies (see the
argument below)), e.g.: $ snakemake --cluster 'qsub
-pe threaded {threads}'.
But, just in case, here is an example of how to use SLURM with anvi-run-workflow:
anvi-run-workflow -w metagenomics \
-c config.json \
--additional-params \
--jobs 10 \
--cluster \
'sbatch --job-name=CHOOSE_A_NICE_JOB_NAME \
--account=YOUR_ACCOUNT \
--output={log} \
--error={log} \
--nodes={threads}'
When you use --cluster, snakemake also requires you to tell it how many jobs may be in the queue at the same time, via --jobs (a.k.a. --cores). From the snakemake help menu:
--cores [N], --jobs [N], -j [N]
Use at most N cores in parallel (default: 1). If N is
omitted, the limit is set to the number of available
cores.
In the example above, --jobs 10 means that at most 10 jobs are submitted to the queue in parallel. Note that this is a count of jobs, not CPUs: if those 10 jobs each request 20 threads, that is 200 CPUs. To cap the total number of threads (CPUs) used across all running jobs, snakemake uses a resource called nodes — every anvi’o workflow rule declares a nodes value equal to its number of threads. anvi’o sets this nodes budget for you automatically, using the max_threads value from your config file, so you normally do not need to pass --resources at all.
If you want to set the total-CPU ceiling explicitly — for example, to a value different from your config’s max_threads — you can pass it yourself with --resources nodes=MAX_THREAD. For instance, adding --resources nodes=48 to --additional-params guarantees that the combined number of threads requested by all simultaneously running jobs never exceeds 48 (so if you have 3 jobs each requiring 20 threads, only two would run in parallel). An explicit --resources value always takes precedence over the one anvi’o would otherwise derive from max_threads. One caveat: for the automatic behavior to kick in, let anvi’o manage --cores for you (i.e. use --jobs as in the example above rather than passing your own --cores); if you pass --cores yourself, anvi’o assumes you are managing resources manually and will not auto-set nodes.
How to use metaSPAdes for assembly
As of anvi’o v5.3 metaSPAdes has been added to the metagenomics workflow. By default, these are the parameters for metaspades:
(...)
"metaspades": {
"additional_params": "--only-assembler",
"threads": 11,
"run": "",
"use_scaffolds": ""
},
(...)
additional_params works in the same way as is explained above for samtools, and allows you to specify anything that metaSPAdes accepts. By default it is set to --only-assembler, since QC is done using iu-filter-quality-minoche, and we see no reason to have metaSPAdes do another step of QC. If you want to specify more parameters then you probably want it to still include --only-assembler.
metaSPAdes has two outputs, contigs.fasta, and scaffolds.fasta. By default anvi’o will use contigs.fasta for the rest of the workflow, but if you want to use scaffolds.fasta, then set use_scaffolds: true in your config file. In any case, anvi’o will save the one you don’t use as well (i.e. by default you will find in your 02_FASTA directory the scaffold.fasta file, and if you choose to use the scaffolds, then you will still find contigs.fasta).
Edit this file to update this information.