Tara Oceans + anvi’o: The story behind Mirusviruses

Table of Contents
- Standing on the shoulders of two giants with Evolution for guidance
- At the interface between Tara Oceans and anvi’o
- Genome-resolved metagenomics for Bacteria, Archaea and Eukarya
- An epiphany on how to navigate metagenomic assemblies using Evolution
- Genome-resolved metagenomics for giant viruses and the mirusviruses
- Towards a decentralized genome-resolved metagenomic journey
Summary
This blog post covers nearly one decade of genome-resolved metagenomic surveys for the sunlit oceans that ultimately led to the discovery of mirusviruses. It tells the story of an epiphany on how Evolution could be used to guide the manual exploration of known and unknown compartments of the marine microbial Tree of Life with metagenomics. The trick was to stand on the shoulders of Tara Oceans and anvi’o with a phylogenetic compass at hand. It was like surveying an immense beach with a metal detector. We found an unusual phylogenetic signal that guided the recovery of large eukaryotic virus genomes forming their very own phylum at the cross-road between two realms. If this blog post had a single objective, it would be to stress the relevance of phylogeny-guided genome-resolved metagenomics in the context of constrained binning and the anvi’o metabins. This relatively simple methodological cocktail can be used to go after environmental genomes of interest in the tsunami of metagenomic data with relative ease.

Standing on the shoulders of two giants with Evolution for guidance
With limited expertise in virology and evolution, by all logic my scientific journey as a microbial ecologist should have remained away from any discoveries contributing to our understanding of virus evolution. I have specialized over the years on environmental genomics with a focus on marine bacterial and eukaryotic populations. But in 2019, I briefly turned my attention to the fascinating world of giant viruses, which triggered a series of fortunate events that ultimately led to the description of a previously unnoticed viral phylum at the crossroad of evolution: the mirusviruses. These viruses are abundant at the surface of the oceans and seas, yet they have eluded scientists’ attention in previous decades in part due to highly unusual evolutionary properties now described in the journal Nature. The only reason I ended up coordinating this scientific adventure is because I have been standing for nearly a decade on the shoulders of two giants: the Tara Oceans expeditions (‘omics data legacy) and bioinformatic platform anvi’o (‘omics data processing and visualization). Together, these two endeavors offer a global view of plankton genomics in high-resolution, with now the ability to zoom in and out on known and unknown compartments of the marine microbial Tree of Life with relative ease. It is like having access to a Google Maps application to explore plankton in various geographic locations and at different levels of resolution: the entire community, microbial lineages, individual genomes, functions, genes, and even single nucleotide variants. This blog explains how we detected and characterized the genomic content of mirusviruses by zooming in on the Tara Oceans metagenomic unknowns using Evolution for guidance.
At the interface between Tara Oceans and anvi’o
The Tara Oceans expeditions provide access to the diversity and functioning of abundant planktonic lineages. Starting in 2009, the consortium behind these expeditions collected planktonic samples across the oceans and seas (Figure 1), which were fractionated by cellular size ranges prior to deeply sequencing their DNA and RNA molecules. This unique metagenomic and metatranscriptomic legacy was made publicly available in 2015 and remains at the center of intense investigations to better understand plankton. Meanwhile, the bioinformatic platform anvi’o provides access to advanced bioinformatic programs and visualization tools for microbial ecologists and evolutionary biologists to manipulate and learn from ‘omics data, without the need for coding expertise. I did not take part in the critical early years of Tara Oceans. On the other hand, I had the chance to witness first-hand how anvi’o metamorphosed from simple ideas to complex lines of code during its early days, at the beautiful Marine Biological Laboratory just a few meters away from the Atlantic Ocean. ") and I were postdocs at the time. I was good with card tricks (from another life) but had a lot to learn about plankton (still do). Since its very first release in 2015, anvi’o has provided the means to perform genome-resolved metagenomics (the recovery of environmental genomes from metagenomic data) in a manual mode using an original interactive interface. The programs and interface provide a unique perspective to explore metagenomic assemblies in the context of environmental signal (metagenomic read recruitment, which informs on the coverage of genomic fragments across samples), allowing the genomic characterization and manual curation of a wide range of microbial populations.
and I were postdocs at the time. I was good with card tricks (from another life) but had a lot to learn about plankton (still do). Since its very first release in 2015, anvi’o has provided the means to perform genome-resolved metagenomics (the recovery of environmental genomes from metagenomic data) in a manual mode using an original interactive interface. The programs and interface provide a unique perspective to explore metagenomic assemblies in the context of environmental signal (metagenomic read recruitment, which informs on the coverage of genomic fragments across samples), allowing the genomic characterization and manual curation of a wide range of microbial populations.
The Tara Oceans ‘omics legacy was made open access just after anvi’o became fully operational on the front of environmental genomics. The release of this considerable amount of data, far from being fully digested by the Tara Oceans consortium, was a rare gift that changed the course of many scientific journeys, including mine.
Genome-resolved metagenomics for Bacteria, Archaea and Eukarya
Together, Tara Oceans and anvi’o provide an effective framework to characterize microbial genomes abundant at the surface of the oceans and seas without the need for cultivation. And so, without having to navigate across the oceans, and without having to learn how to code for any programing language, I started processing the Tara Oceans metagenomes to extract environmental genomes. In two sequential surveys carried on at the University of Chicago with Meren (2015/2016 – size fraction enriched in bacteria), and Genoscope in France with Olivier Jaillon, Eric Pelletier and other researchers already involved in the early days of Tara Oceans (2017/2018 – size fractions enriched in eukaryotes), we performed large metagenomic co-assemblies one oceanic region at a time. The Figure 1 describes those regions for the second survey. These assemblies produced more than 10 million genomic fragments, commonly referred to as contigs. Each contig represents one piece of a complex puzzle. In theory, making sense of the puzzle should provide a genomic context for most of the main planktonic lineages. In the case of Bacteria, various genes are known, based on our remarkable culture portfolio, to be present one time in almost every genome. Thus, bacterial genomes are recognizable by the occurrence of these single copy core genes (thereafter called marker genes for simplicity) among the assembled contigs. In practice, one can identify environmental bacterial genomes within the large metagenomic assemblies using two key information. First, contigs from the same bacterial population tend to correlate across the Tara Oceans samples because the corresponding molecules occur in similar amount within each cell. Second, contigs from the same bacterial population have a complement of marker genes. The exact same approach can be applied to Archaea and eukaryotes based on other sets of marker genes.
Since anvi’o contains marker gene collections for the three domains of life, I could use the platform to characterize and manually curate a substantial number of environmental genomes for Bacteria, Archaea and the eukaryotes. Anvi’o cannot visualize more than about 30,000 contigs at a time, so the trick was to first perform a step of constrained binning to produce self-sustained anvi’o metabins. By design, each metabin can contain multiple genomes that are often completely unrelated in term of taxonomy. These two genome-resolved metagenomic surveys required many months of laborious manual binning by systematically exploring thousands of metabins in the anvi’o interactive interface. They lacked sophistication (e.g., rudimentary taxonomic perspective) and were painful to complete. But prior to walking with confidence, a child first needs to learn how to crawl in the ground. And indeed, a lot of experience was gained when completing these two surveys. In particular, the interface was insightful and critical in assessing contig correlation patterns across samples and gain confidence in the quality of environmental genomes. The Asgard (archaeal superphylum) example displayed in the Figure 1 conveys that important message. The Figure illustrates the manual characterization (panel B) and curation (panel C) of an Asgard environmental genome from one Indian Ocean metabin (the data is available for teaching and training purposes), with layers displaying their environmental signal (mean coverage) across Tara Oceans samples within that region. The clear signal of contig coverage values across samples and complement of marker genes strongly support the biological relevance of this environmental genome. In the end, not only did the two surveys provide some insights into the genomics of various planktonic cellular lineages (e.g., abundant and widespread Trichodesmium species that lost the ability to fix nitrogen, or else the 1.3 Gb long genome of a diatom population that recurrently blooms in the Southern Ocean), but they also provided the conceptual and methodological foundation that would help us walk confidently towards the mirusviruses. However, what I needed first was an epiphany on how Evolution would guide our genomic exploration of metagenomic assemblies.
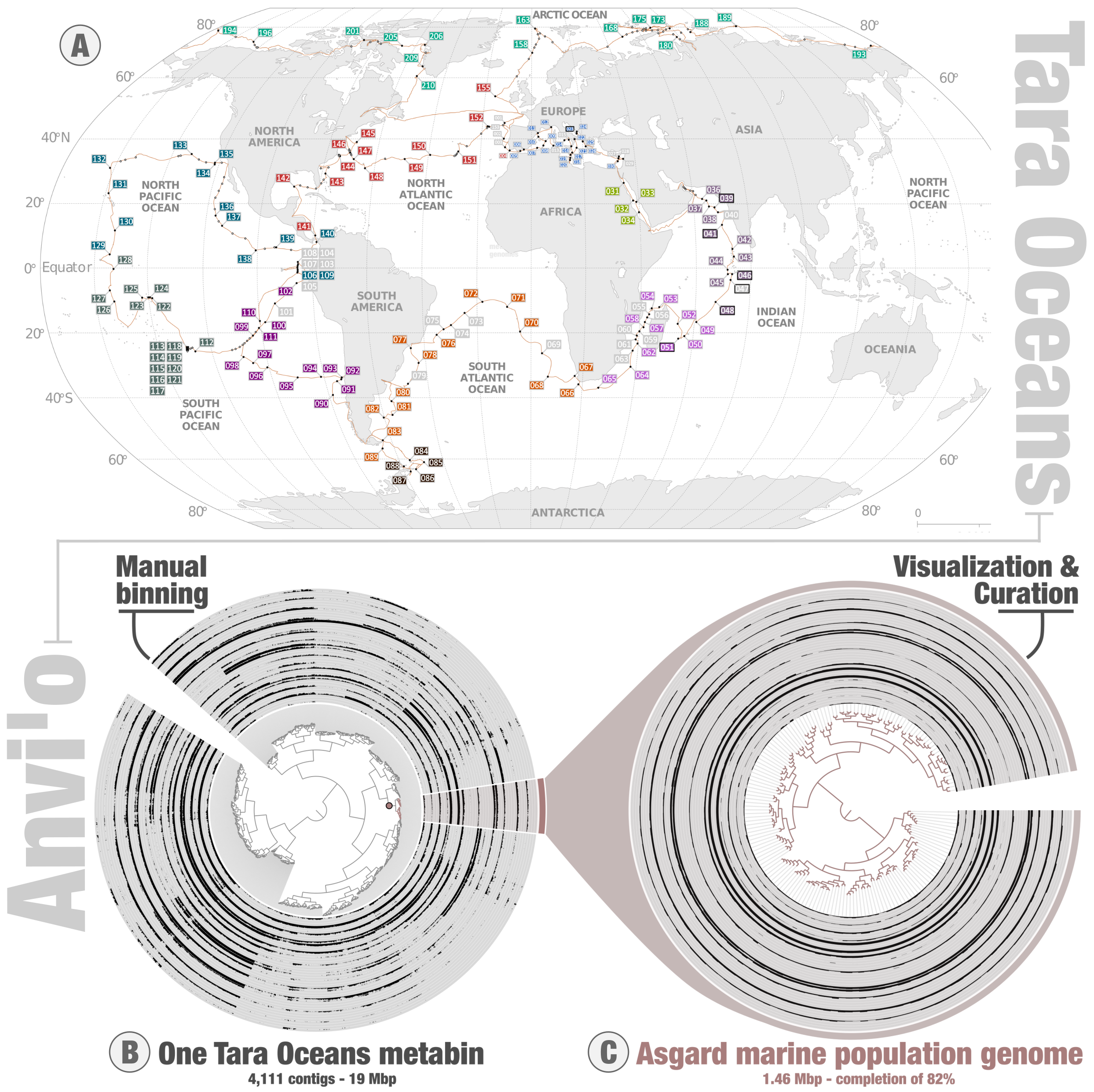
Figure 1: Tara Oceans and the anvi’o metabins. Panel A describes the location of Tara Oceans stations, colored by regions used for the large metagenomic co-assemblies. Panel B describes the anvi’o interactive interface for genome-resolved metagenomics, applied to one Tara Oceans metabin of ~4k contigs organized based on their differential coverage and sequence composition. Panel C corresponds to the manual curation of a subset of contigs from that metabin, which corresponds to an environmental genome of Asgard Archaea. Contigs are organized based on their sequence composition alone.
An epiphany on how to navigate metagenomic assemblies using Evolution
The first two surveys focused on marine microbial populations from the three domains of life, overlooking the many viruses infecting them. But just before securing a lifetime research position in France (CNRS), I completed a very short postdoc experience with Patrick Forterre at the Pasteur Institute (2019). Patrick is an expert in virology (e.g., concept of virocell) with a particular interest in the early life on Earth. After many years studying the ecology of microorganisms, I had entered a temple for Evolution. There, I was introduced by Patrick and Morgan Gaïa (a long-term postdoc in his lab that is now a close colleague at the Genoscope) to the evolutionary wonders of the DNA-dependent RNA polymerases. These polymerases are at the center of transcription mechanisms by synthesizing RNA from DNA molecules. Genes encoding these proteins are long, evolutionary constrained (meaning they evolve relatively slowly over long periods of time) and are rarely transferred between distantly related clades. There are two subunits: RNApolA and RNApolB. Not only are their corresponding genes present in every bacterial, archaeal and eukaryotic genome, but they also occur in most giant virus genomes. At the time of my visit, Morgan, Patrick, and their colleagues had just demonstrated using in part these two subunits that giant viruses predate the origin of modern eukaryotes. As part of their study, they had generated hidden Markov models to identify distantly related RNApolA and RNApolB proteins and also created reference databases for these markers covering most clades known from cultivation.
On the one side, Morgan and Patrick had an effective framework to place DNA-dependent RNA polymerases in the Tree of Life, within and behyond the three domains of life. On the other side, I had thousands of anvi’o metabins that cover plankton genomics from pole to pole. However, I did not have any elaborate ways to determine what kind of environmental genomes the metabins contained prior to binning them manually. Looking at both sides, it became evident that markers such as the DNA-dependent RNA polymerases could provide an effective bridge to connect Evolution and genome-resolved metagenomics. Indeed, in theory the RNApolA and RNApolB could not only point to a wide range of environmental genomes across the metabins, but they could also tell us with hight precision their placement in the Tree of Life. If I only wanted to explore the genus Trichodesmium, I would simply need to find DNA-dependent RNA polymerases matching to this clade and jump into their corresponding metabins to extract the focal environmental genomes. Perhaps more importantly, if there were entirely new clades of RNApolA or RNApolB compared to the references, I could also go after them. A simple blast against reference databases could even provide a novelty score for each RNApolA and RNApolB gene. The lower the identity score, the higher the novelty. In other words, the DNA-dependent RNA polymerases could provide the guidance I needed to effectively explore known amd unknown compartments of the Tara Oceans ‘omics legacy with ease. This was an epiphany regarding how Evolution could be used as a compass for genome-resolved metagenomics. From that moment, I stopped crawling, and started walking confidently among the metabins and within the complexity of plankton genomics at a global scale.
Genome-resolved metagenomics for giant viruses and the mirusviruses
At Pasteur, we screened for the RNApolB among the Tara Oceans metagenomes to look specifically for giant viruses. A phylogenetic tree was inferred using all the diversity of RNApolB found in the large Tara Oceans metagenomic co-assemblies, plus some references from culture for perspective. Major clades emerged for Bacteria, Archaea, eukaryotes, and the different giant virus clades already known from culture (Figure 2). In addition, we could observe few deep-branching RNApolB clades lacking any known references (referred to as “new” in the figure). Prior to this screening, I remember us expecting to find a few dozen hits for giant viruses. Instead, we got thousands of hits, and realized a bit late (we got scooped not just once but twice by our esteemed peers overseas) that the data was a gold mine to extract environmental genomes for the giant viruses. Of course, we were also captivated by the mysterious deep-branching RNApolB clades. Our view was that they most likely represented new kinds of giant viruses. However, anything was possible, and plasmids for example could not be ruled out from that perspective alone. What we needed was a genomic context. And so, still standing on the shoulders of Tara Oceans and anvi’o, I started a third genome-resolved metagenomic survey, but this time with a phylogenetic twist in it. After years of metagenomic explorations with no evolutionary perspective, it was like turning on the lights and finally seeing the extent of planktonic diversity within and beyond the microbial cells with great clarity. This was genome-resolved metagenomics on steroids.
Instead of exploring each metabin one by one (as was done for the two first surveys), this time I could start with the phylogenetic signal and focus solely on metabins containing RNApolB clades of interest (Figure 2). Not only did I gain a considerable amount of time, but I could also turn my attention to the phylogenetic signal displaying the highest novelty (the deep-branching new RNApolB clades). Using contigs containing the RNApolB genes of interest, I exploited the anvi’o interactive interface to search for other contigs they might correlate with. In many cases, we could observe a strong association between the targeted contig and others, forming putative environmental genomes (see example in Figure 2C). Like in the example of the Asgard environmental genome, being able to observe these patterns was critical. Once again, the interface gave us strong confidence in the environmental signal. This third genome-resolved metagenomic survey was a blast. It dragged me into the fascinating world of marine DNA viruses with the guidance of Morgan, initiated productive collaborations (e.g., with Hiroyuki Ogata, Lingjie Meng and Mart Krupovic), expanded the known genomic diversity of giant viruses, and led to an unexpected discovery. After many months of genomic investigations, including a major twist with the recovery of an unexpected major capsid protein by Mart, our international research team could demonstrate that most of the new RNApolB clades correspond to a previously unnoticed phylum of eukaryotic DNA viruses displaying chimeric attributes in between giant viruses and herpesviruses: the mirusviruses.
Mirusvirus genomes characterized thus far represent just 0.004% of metagenomic reads from Tara Oceans. It is a needle in a haystack. But with guidance from the RNApolB phylogeny, we only had to perform genome-resolved metagenomics on 3% of the produced anvi’o metabins to go after them. The entire process was like surveying an immense beach with a highly sensitive metal detector. We were looking for giant viruses, but instead we found one of the many evolutionary treasures occuring whithin the complexity of plankton.
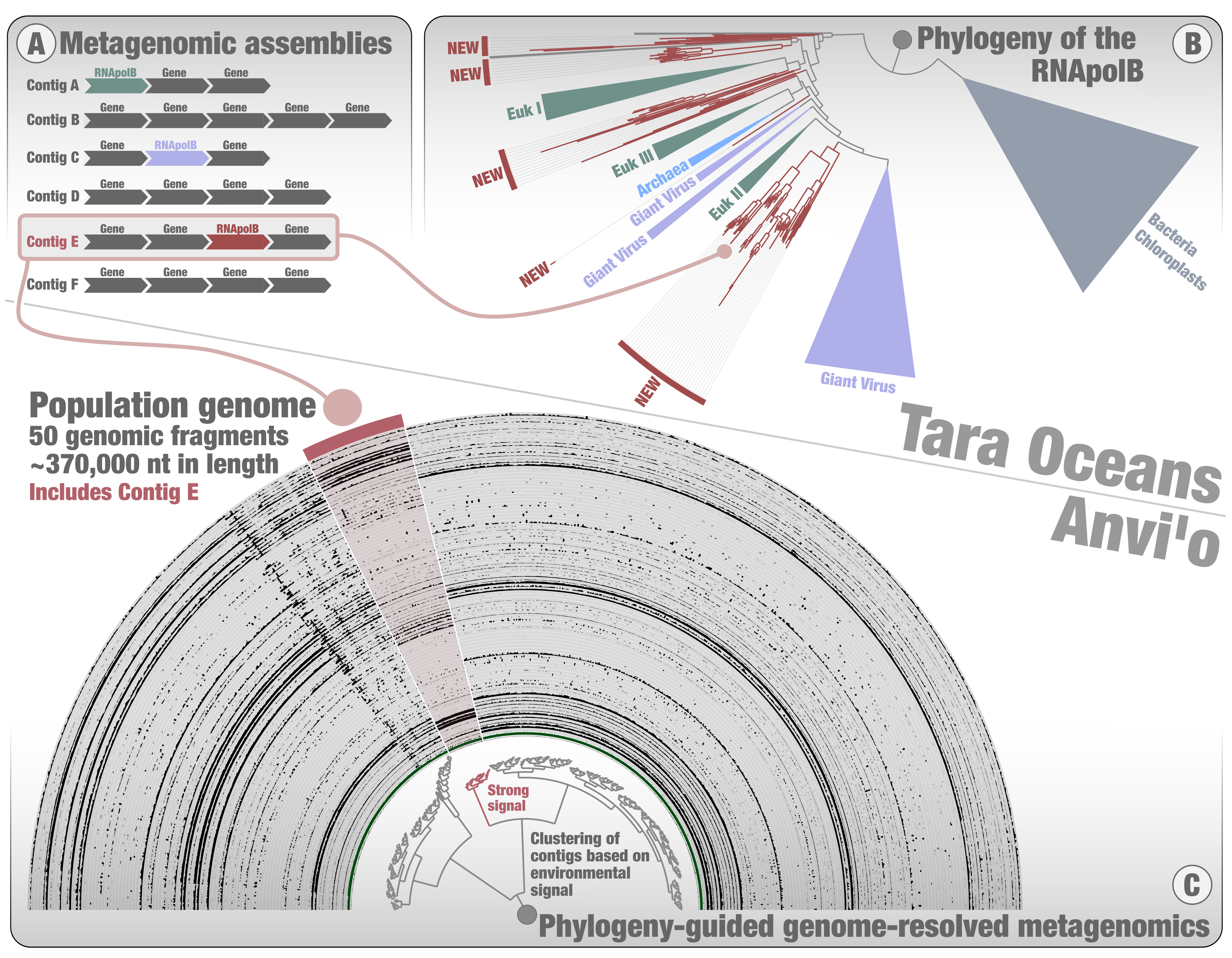
Figure 2: A summary of phylogeny-guided genome-resolved metagenomics with the anvi’o metabins. The figure displays the phylogeny of DNA-dependent RNA polymerase subunit B (RNApolB) found in the large Tara Oceans metagenomic assemblies and exemplifies how contigs containing RNApolB genes of interest can be used to identify putative environmental genomes among the metabins, using the anvi’o interactive interface.
Towards a decentralized genome-resolved metagenomic journey
It is now 2023. I have lost most of my skills in card tricks (along with my hair), but my understanding of plankton genomics has developed in unexpected ways. Realizing how the DNA-dependent RNA polymerases could be used to explore the large Tara Oceans metagenomic co-assemblies was a pivotal moment. This lucky event in my scientific journey followed others. First, my PhD mentors Tim Vogel and Pascal Simonet at the Ecole Central de Lyon pointed me towards metagenomics early on. Then, Tara Oceans and anvi’o gave me a unique metagenomic legacy and the bioinformatic superpowers to explore it. And finally, Patrick and Morgan gave me an evolutionary compass to explore a large fraction of the marine microbial Tree of Life that goes well behond Bacteria, Archaea and Eukarya. That latest gift unlocked the discovery of mirusviruses and made me realize that there was another way to perform genome-resolved metagenomics. Not blindly like in the two first surveys, but instead using phylogenetic signal as a compass to immediately go after clades of interest, as done in the third survey. This methodology can be referred to as “phylogeny-guided genome-resolved metagenomics”. The main concept behind this methodology is not necessarily novel (e.g., tools exist to screeen metagenomic assemblies for phylogenetic signal of interest), and it falls within the broader scope of targeted binning that can also use other types of signal (e.g., a function of interest by  ) for guidance. Thus, this blog is not about a new methodology or concept, but rather about how relevant phylogenetic signal was used in the context of anvi’o metabins to characterize interesting environmental genomes.
) for guidance. Thus, this blog is not about a new methodology or concept, but rather about how relevant phylogenetic signal was used in the context of anvi’o metabins to characterize interesting environmental genomes.
Constrained binning and the anvi’o metabins are particularely relevant to navigate among large metagenomic assemblies regardless of the metagenomic signal used for guidance (e.g., phylogeny, taxonomy, function, complete metabolic pathway, or even a combination of pathways - your imagination is the limit!). Each metabin can be visualized in the anvi’o interactive interface to manually delineate environmental genomes of known and unknown origin with confidence, as examplified in the two figures. In the case of Tara Oceans and the RNApolB marker gene, our phylogenetic tree pointed to a small forest of metabins containing large and complex environmental genomes distant from anything known before. But what can other markers reveal within the scope of Tara Oceans? And what awaits to be discovered in other ecosystems? Expanding this approach to other markers and ecosystems is already providing more insights. Critically, decorating metabins with relevant signal provides the ability to zoom in and out on known and unknown compartments of metagenomics with relative ease. This methodology could play a more important role in the ongoing efforts to characterize the microbial Tree of Life with environmental genomics, however some bottlenecks need to be resolved first if we want to democratize its use among microbial ecologists and evolutionary biologists. This is a work in progress. Maybe I am an idealist, but I am looking towards a decentralized genome-resolved metagenomic journey that would empower cohorts of motivated researchers to manually characterize, curate and exploit a broad range of environmental genomes from large metagenomic projects. If this were to work, no one would have to perform large genome-resolved metagenomic surveys on their own. Instead, we could explore the environmental genomics of microorganisms and their viruses together!