Help pages for anvi'o programs and artifacts
Table of Contents
Here you will find a list of all anvi’o programs and artifacts that enable constructing workflows for integrated multi ‘omics investigations.
If you need an introduction to the terminology used in ‘omics research or in anvi’o, please take a look at our vocabulary page. The anvi’o community is with you! If you have practical, technical, or science questions this page to learn about resources available to you. If you are feeling overwhelmed, you can always scream towards the anvi’o 
The help contents were last updated on 17 Mar 26 09:37:20 for anvi’o version 9-dev (eunice).
The latest version of anvi’o is v9. See the release notes.
Anvi’o workflows
Anvi’o workflows are dynamic recipes for easy-to-use, scalable, and reproducible bioinformatics analyses through orchestrated use of anvi’o programs as well as third-party software. These workflows typically start with raw data files and a workflow-config ![]() and produce anvi’o artifacts, which enable you to outsource rudimentary and relatively well-understood initial steps of your ‘omics analyses so you can focus on more critical downstream research questions by further analyzing these data products inside or outside of the anvi’o software ecosystem.
and produce anvi’o artifacts, which enable you to outsource rudimentary and relatively well-understood initial steps of your ‘omics analyses so you can focus on more critical downstream research questions by further analyzing these data products inside or outside of the anvi’o software ecosystem.
The anvi’o 9-dev (eunice) contains 5 workflows:
- The anvi'o contigs workflow by


") | From FASTA files to annotated anvi'o contigs databases.
| From FASTA files to annotated anvi'o contigs databases.
- The anvi'o metagenomics workflow by | From FASTA and/or FASTQ files to anvi'o contigs and profile databases.
- The anvi'o ecophylo workflow by | Co-characterize the biogeography and phylogeny of any protein.
- The anvi'o trnaseq workflow by | Process transfer RNA transcripts from tRNA-seq datasets.

- The anvi'o sra-download workflow by | Download, verify, extract, and gzip paired-end FASTQ files automatically from the NCBI short-read archive (SRA).
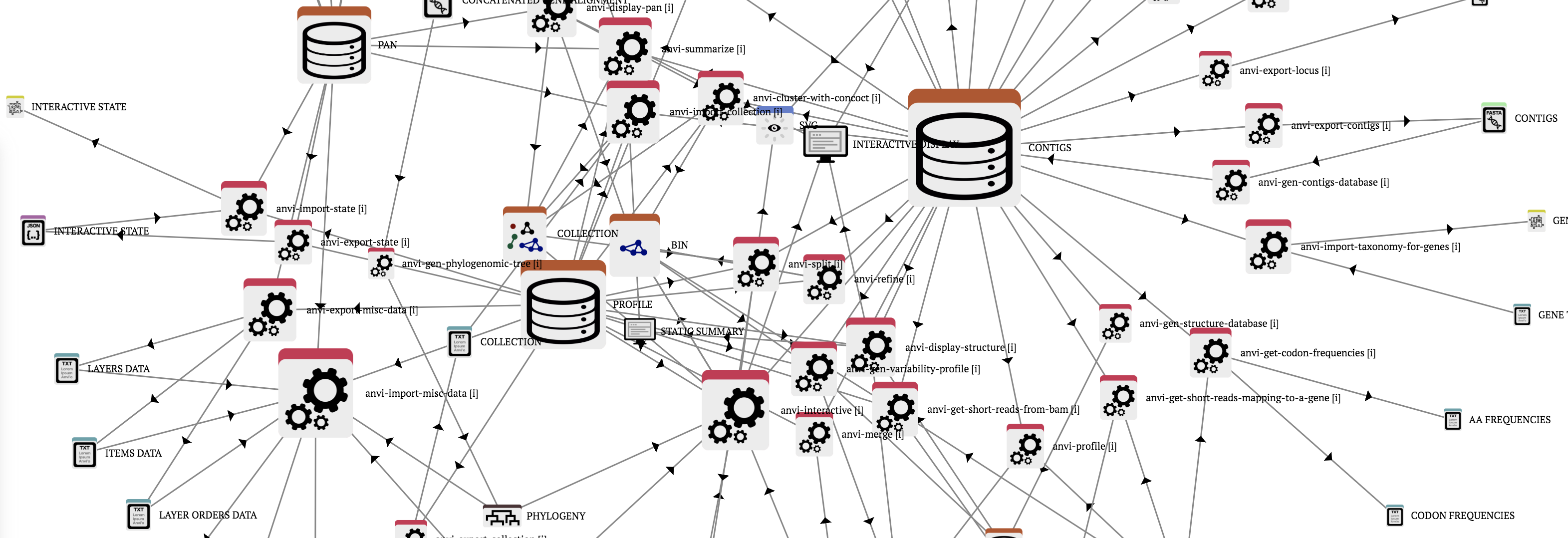
Anvi’o artifacts
Anvi’o artifacts represent concepts, file types, or data types anvi’o programs can work with. A given anvi’o artifact can be provided by the user (such as a FASTA file), produced by anvi’o (such as a profile database), or both (such as phylogenomic trees). Anvi’o artifacts link anvi’o programs to each other to build novel workflows.
Listed below a total of 143 artifacts.
Anvi’o programs
Anvi’o programs perform atomic tasks that can be weaved together to implement complete ‘omics workflows. Please note that there may be programs that are not listed on this page. You can type ‘anvi-‘ in your terminal, and press the TAB key twice to see the full list of programs available to you on your system, and type anvi-program-name --help to read the full list of command line options.
Listed below a total of 160 programs.
| 🔥 anvi-analyze-synteny. Extract ngrams, as in 'co-occurring genes in synteny', from genomes. |
|
🧀
genomes-storage-db |
|
🍕
ngrams |
|
🧠 |
| 🔥 anvi-cluster-contigs. A program to cluster items in a merged anvi'o profile using automatic binning algorithms. |
|
🧀
profile-db |
|
🍕
collection |
🧠  |
| 🔥 anvi-compute-completeness. A script to generate completeness info for a given list of splits. |
|
🧀
contigs-db |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-compute-functional-enrichment-across-genomes. A program that computes functional enrichment across groups of genomes.. |
|
🧀
groups-txt |
|
🍕
functional-enrichment-txt |
🧠  |
| 🔥 anvi-compute-functional-enrichment-in-pan. A program that computes functional enrichment within a pangenome.. |
|
🧀
misc-data-layers |
|
🍕
functional-enrichment-txt |
|
🧠 |
| 🔥 anvi-compute-gene-cluster-homogeneity. Compute homogeneity for gene clusters. |
|
🧀
pan-db |
| 🍕 n/a |
🧠  |
| 🔥 anvi-compute-genome-similarity. Export sequences from sequence sources and compute a similarity metric (e.g. ANI). If a Pan Database is given anvi'o will write computed output to misc data tables of Pan Database. |
|
🧀
external-genomes |
|
🍕
genome-similarity |
🧠  |
| 🔥 anvi-compute-metabolic-enrichment. A program that computes metabolic enrichment across groups of genomes and metagenomes. |
|
🧀
kegg-metabolism |
|
🍕
functional-enrichment-txt |
|
🧠 |
| 🔥 anvi-compute-rarefaction-curves. A program that computes rarefaction curves and Heaps' Law fit for a given pangenome. |
|
🧀
pan-db |
|
🍕
rarefaction-curves |
🧠  |
| 🔥 anvi-db-info. Access self tables, display values, or set new ones totally on your own risk. |
|
🧀
pan-db |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-delete-collection. Remove a collection from a given profile database. |
|
🧀
profile-db |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-delete-functions. Remove functional annotation sources from an anvi'o contigs database. |
|
🧀
contigs-db |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-delete-hmms. Remove HMM hits from an anvi'o contigs database. |
|
🧀
contigs-db |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-delete-misc-data. Remove stuff from 'additional data' or 'order' tables for either items or layers in either pan or profile databases. OR, remove stuff from the 'additional data' tables for nucleotides or amino acids in contigs databases. |
|
🧀
pan-db |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-delete-state. Delete an anvi'o state from a pan or profile database. |
|
🧀
pan-db |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-dereplicate-genomes. Identify redundant (highly similar) genomes. |
|
🧀
external-genomes |
|
🍕
fasta |
|
🧠 |
| 🔥 anvi-display-contigs-stats. Start the anvi'o interactive interface for viewing or comparing contigs statistics.. |
|
🧀
contigs-db |
|
🍕
contigs-stats |
🧠   |
| 🔥 anvi-display-codon-frequencies. Display codon frequency statistics across genes in a given genome in the anvi'o interactive interface.. |
|
🧀
contigs-db |
|
🍕
interactive |
|
🧠 |
| 🔥 anvi-display-functions. Start an anvi'o interactive display to see functions across genomes. |
|
🧀
functions |
|
🍕
interactive |
|
🧠 |
| 🔥 anvi-display-metabolism. Start the anvi'o interactive interactive for viewing KEGG metabolism data. |
|
🧀
contigs-db |
|
🍕
interactive |
|
🧠 |
| 🔥 anvi-display-pan. Start an anvi'o server to display a pan-genome. |
|
🧀
pan-db |
|
🍕
collection |
|
🧠 |
| 🔥 anvi-display-structure. Interactively visualize sequence variants on protein structures. |
|
🧀
structure-db |
|
🍕
interactive |
|
🧠 |
| 🔥 anvi-draw-kegg-pathways. Write KEGG pathway map files incorporating data sourced from anvi'o databases. |
|
🧀
contigs-db |
|
🍕
kegg-pathway-map |
|
🧠 |
| 🔥 anvi-estimate-genome-completeness. Estimate completion and redundancy using domain-specific single-copy core genes. |
|
🧀
contigs-db |
|
🍕
completion |
|
🧠 |
| 🔥 anvi-estimate-metabolism. Reconstructs metabolic pathways and estimates pathway completeness for a given set of contigs. |
|
🧀
contigs-db |
|
🍕
kegg-metabolism |
|
🧠 |
| 🔥 anvi-estimate-scg-taxonomy. Estimates taxonomy at genome and metagenome level. This program is the entry point to estimate taxonomy for a given set of contigs (i.e., all contigs in a contigs database, or contigs described in collections as bins). For this, it uses single-copy core gene sequences and the GTDB database. |
|
🧀
profile-db |
|
🍕
genome-taxonomy |
🧠  |
| 🔥 anvi-estimate-trna-taxonomy. Estimates taxonomy at genome and metagenome level using tRNA sequences.. |
|
🧀
profile-db |
|
🍕
genome-taxonomy |
|
🧠 |
| 🔥 anvi-experimental-organization. Create an experimental clustering dendrogram.. |
|
🧀
clustering-configuration |
|
🍕
dendrogram |
|
🧠 |
| 🔥 anvi-export-collection. Export a collection from an anvi'o database. |
|
🧀
profile-db |
|
🍕
collection-txt |
|
🧠 |
| 🔥 anvi-export-contigs. Export contigs (or splits) from an anvi'o contigs database. |
|
🧀
contigs-db |
|
🍕
contigs-fasta |
|
🧠 |
| 🔥 anvi-export-functions. Export functions of genes from an anvi'o contigs database for a given annotation source. |
|
🧀
contigs-db |
|
🍕
functions-txt |
|
🧠 |
| 🔥 anvi-export-gene-calls. Export gene calls from an anvi'o contigs database. |
|
🧀
contigs-db |
|
🍕
gene-calls-txt |
|
🧠 |
| 🔥 anvi-export-gene-clusters. Export gene clusters in a pan-db as a three-column, TAB-delimited file that associates each gene call in each genome with a gene cluster. |
|
🧀
pan-db |
|
🍕
gene-clusters-txt |
|
🧠 |
| 🔥 anvi-export-gene-coverage-and-detection. Export gene coverage and detection data for all genes associated with contigs described in a profile database. |
|
🧀
profile-db |
|
🍕
coverages-txt |
|
🧠 |
| 🔥 anvi-export-items-order. Export an item order from an anvi'o database. |
|
🧀
pan-db |
|
🍕
misc-data-items-order-txt |
|
🧠 |
| 🔥 anvi-export-locus. This program helps you cut a 'locus' from a larger genetic context (e.g., contigs, genomes). By default, anvi'o will locate a user-defined anchor gene, extend its selection upstream and downstream based on the –num-genes argument, then extract the locus to create a new contigs database. The anchor gene must be provided as –search-term, –gene-caller-ids, or –hmm-sources. If –flank-mode is designated, you MUST provide TWO flanking genes that define the locus region (Please see –flank-mode help for more information). If everything goes as plan, anvi'o will give you individual locus contigs databases for every matching anchor gene found in the original contigs database provided. Enjoy your mini contigs databases!. |
|
🧀
contigs-db |
|
🍕
locus-fasta |
|
🧠 |
| 🔥 anvi-export-misc-data. Export additional data or order tables in pan or profile databases for items or layers. |
|
🧀
pan-db |
|
🍕
misc-data-items-txt |
|
🧠 |
🔥 anvi-export-splits-and-coverages. Export split or contig sequences and coverages across samples stored in an anvi'o profile database. This program is especially useful if you would like to 'bin' your splits or contigs outside of anvi'o and import the binning results into anvi'o using anvi-import-collection program.
|
|
🧀
profile-db |
|
🍕
contigs-fasta |
|
🧠 |
| 🔥 anvi-export-splits-taxonomy. Export taxonomy for splits found in an anvi'o contigs database. |
|
🧀
contigs-db |
|
🍕
splits-taxonomy-txt |
|
🧠 |
| 🔥 anvi-export-state. Export an anvi'o state into a profile database. |
|
🧀
pan-db |
|
🍕
state-json |
|
🧠 |
| 🔥 anvi-export-structures. Export .pdb structure files from a structure database. |
|
🧀
structure-db |
|
🍕
protein-structure-txt |
|
🧠 |
| 🔥 anvi-gen-contigs-database. Generate a new anvi'o contigs database. |
|
🧀
contigs-fasta |
|
🍕
contigs-db |
|
🧠 |
| 🔥 anvi-gen-fixation-index-matrix. Generate a pairwise matrix of a fixation indices between samples. |
|
🧀
contigs-db |
|
🍕
fixation-index-matrix |
|
🧠 |
| 🔥 anvi-gen-gene-consensus-sequences. Collapse variability for a set of genes across samples. |
|
🧀
profile-db |
|
🍕
genes-fasta |
|
🧠 |
🔥 anvi-gen-gene-level-stats-databases. A program to compute genes databases for a ginen set of bins stored in an anvi'o collection. Genes databases store gene-level coverage and detection statistics, and they are usually computed and generated automatically when they are required (such as running anvi-interactive with --gene-mode flag). This program allows you to pre-compute them if you don't want them to be done all at once.
|
|
🧀
profile-db |
|
🍕
genes-db |
|
🧠 |
| 🔥 anvi-gen-genomes-storage. Create a genome storage from internal and/or external genomes for a pangenome analysis. |
|
🧀
external-genomes |
|
🍕
genomes-storage-db |
|
🧠 |
| 🔥 anvi-gen-phylogenomic-tree. Generate phylogenomic tree from aligment file. |
|
🧀
concatenated-gene-alignment-fasta |
|
🍕
phylogeny |
|
🧠 |
| 🔥 anvi-gen-structure-database. Creates a database of protein structures. Predict protein structures using template-based homology modelling of genes in your contigs database, or import pre-computed PDB structures you already have.. |
|
🧀
contigs-db |
|
🍕
structure-db |
|
🧠 |
| 🔥 anvi-gen-variability-network. Generate a network description from an anvi'o variability profile.. |
|
🧀
variability-profile-txt |
|
🍕
variability-profile-xml |
|
🧠 |
| 🔥 anvi-gen-variability-profile. Generate a table that comprehensively summarizes the variability of nucleotide, codon, or amino acid positions. We call these single nucleotide variants (SNVs), single codon variants (SCVs), and single amino acid variants (SAAVs), respectively. |
|
🧀
contigs-db |
|
🍕
variability-profile-txt |
|
🧠 |
| 🔥 anvi-get-aa-counts. Fetches the number of times each amino acid occurs from a contigs database in a given bin, set of contigs, or set of genes. |
|
🧀
splits-txt |
|
🍕
aa-frequencies-txt |
|
🧠 |
| 🔥 anvi-get-codon-frequencies. Get codon or amino acid frequency statistics from genomes, genes, and functions. |
|
🧀
contigs-db |
|
🍕
codon-frequencies-txt |
|
🧠 |
| 🔥 anvi-get-codon-usage-bias. Get codon usage bias (CUB) statistics of genes and functions. |
|
🧀
contigs-db |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-get-metabolic-model-file. This program exports a metabolic reaction network to a file suitable for flux balance analysis.. |
|
🧀
contigs-db |
|
🍕
reaction-network-json |
|
🧠 |
| 🔥 anvi-get-pn-ps-ratio. Calculate the rates of non-synonymous and synonymous polymorphism for genes across environmetns using the output of anvi-gen-variability-profile.. |
|
🧀
contigs-db |
|
🍕
pn-ps-data |
|
🧠 |
| 🔥 anvi-get-sequences-for-gene-calls. A script to get back sequences for gene calls. |
|
🧀
contigs-db |
|
🍕
genes-fasta |
|
🧠 |
| 🔥 anvi-get-sequences-for-gene-clusters. Do cool stuff with gene clusters in anvi'o pan genomes. |
|
🧀
pan-db |
|
🍕
genes-fasta |
|
🧠 |
| 🔥 anvi-get-sequences-for-hmm-hits. Get sequences for HMM hits from many inputs. |
|
🧀
contigs-db |
|
🍕
genes-fasta |
|
🧠 |
| 🔥 anvi-get-short-reads-from-bam. Get short reads back from a BAM file with options for compression, splitting of forward and reverse reads, etc. |
|
🧀
profile-db |
|
🍕
short-reads-fasta |
|
🧠 |
| 🔥 anvi-get-short-reads-mapping-to-a-gene. Recover short reads from BAM files that were mapped to genes you are interested in. It is possible to work with a single gene call, or a bunch of them. Similarly, you can get short reads from a single BAM file, or from many of them. |
|
🧀
contigs-db |
|
🍕
short-reads-fasta |
|
🧠 |
| 🔥 anvi-get-split-coverages. Export splits and the coverage table from database. |
|
🧀
profile-db |
|
🍕
coverages-txt |
|
🧠 |
| 🔥 anvi-get-tlen-dist-from-bam. Report the distribution of template lengths from a BAM file. The purpose of this is to get an idea about the insert size distribution in a BAM file rapidly by summarizing distances between each paired-end read in a given read recruitment experiment.. |
|
🧀
bam-file |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-import-collection. Import an external binning result into anvi'o. |
|
🧀
contigs-db |
|
🍕
collection |
|
🧠 |
| 🔥 anvi-import-functions. Parse and store functional annotation of genes. |
|
🧀
contigs-db |
|
🍕
functions |
|
🧠 |
| 🔥 anvi-import-items-order. Import a new items order into an anvi'o database. |
|
🧀
pan-db |
|
🍕
misc-data-items-order |
|
🧠 |
| 🔥 anvi-import-metabolite-profile. This program imports metabolite abundance data and stores it in a profile database.. |
|
🧀
profile-db |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-import-misc-data. Populate additional data or order tables in pan or profile databases for items and layers, OR additional data in contigs databases for nucleotides and amino acids (the Swiss army knife-level serious stuff). |
|
🧀
pan-db |
|
🍕
misc-data-items |
|
🧠 |
| 🔥 anvi-import-protein-profile. This program imports protein abundance data into a profile database.. |
|
🧀
profile-db |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-import-state. Import an anvi'o state into a profile database. |
|
🧀
pan-db |
|
🍕
state |
|
🧠 |
| 🔥 anvi-import-taxonomy-for-genes. Import gene-level taxonomy into an anvi'o contigs database. |
|
🧀
contigs-db |
|
🍕
gene-taxonomy |
|
🧠 |
| 🔥 anvi-import-taxonomy-for-layers. Import layers-level taxonomy into an anvi'o additional layer data table in an anvi'o single-profile database. |
|
🧀
single-profile-db |
|
🍕
layer-taxonomy |
|
🧠 |
| 🔥 anvi-init-bam. Sort/Index BAM files. |
|
🧀
raw-bam-file |
|
🍕
bam-file |
|
🧠 |
| 🔥 anvi-inspect. Start an anvi'o inspect interactive interface. |
|
🧀
profile-db |
|
🍕
interactive |
|
🧠 |
| 🔥 anvi-interactive. Start an anvi'o server for the interactive interface. |
|
🧀
profile-db |
|
🍕
collection |
🧠   |
| 🔥 anvi-matrix-to-newick. Takes a distance matrix, returns a newick tree. |
|
🧀
view-data |
|
🍕
dendrogram |
|
🧠 |
| 🔥 anvi-merge. Merge multiple anvio profiles. |
|
🧀
single-profile-db |
|
🍕
profile-db |
|
🧠 |
| 🔥 anvi-merge-bins. Merge a given set of bins in an anvi'o collection. |
|
🧀
pan-db |
| 🍕 n/a |
|
🧠 |
🔥 anvi-merge-trnaseq. This program processes one or more anvi'o tRNA-seq databases produced by anvi-trnaseq and outputs anvi'o contigs and merged profile databases accessible to other tools in the anvi'o ecosystem. Final tRNA "seed sequences" are determined from a set of samples. Each sample yields a set of tRNA predictions stored in a tRNA-seq database, and these tRNAs may be shared among the samples. tRNA may be 3' fragments and thereby subsequences of longer tRNAs from other samples which would become seeds. The profile database produced by this program records the coverages of seeds in each sample. This program finalizes predicted nucleotide modification sites using tunable substitution rate parameters.
|
|
🧀
trnaseq-db |
|
🍕
trnaseq-contigs-db |
|
🧠 |
| 🔥 anvi-meta-pan-genome. Convert a pangenome into a metapangenome. |
|
🧀
internal-genomes |
|
🍕
metapangenome |
|
🧠 |
| 🔥 anvi-migrate. Migrates any anvi'o artifact, whether it is a database or a config file, to a newer version. Pure magic.. |
|
🧀
contigs-db |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-oligotype-linkmers. Takes an anvi'o linkmers report, generates an oligotyping output. |
|
🧀
linkmers-txt |
|
🍕
oligotypes |
|
🧠 |
| 🔥 anvi-pan-genome. An anvi'o program to compute a pangenome from an anvi'o genome storage. |
|
🧀
genomes-storage-db |
|
🍕
pan-db |
|
🧠 |
| 🔥 anvi-plot-trnaseq. A program to write plots of coverage and modification data from flexible groups of tRNA-seq seeds. |
|
🧀
trnaseq-contigs-db |
|
🍕
trnaseq-plot |
| 🧠 |
| 🔥 anvi-predict-metabolic-exchanges. Predicts metabolic exchanges between microbes based on the reaction network. |
|
🧀
contigs-db |
|
🍕
metabolite-exchange-predictions |
|
🧠 |
🔥 anvi-profile. The flagship anvi'o program to profile a BAM file. Running this program on a BAM file will quantify coverages per nucleotide position in read recruitment results and will average coverage and detection data per contig. It will also calculate single-nucleotide, single-codon, and single-amino acid variants, as well as structural variants, such as insertion and deletions, to eventually stores all data into a single anvi'o profile database. For very large projects, this program can demand a lot of time, memory, and storage resources. If all you want is to learn coverages of your nutleotides, genes, contigs, or your bins collections from BAM files very rapidly, and/or you do not need anvi'o single profile databases for your project, please see other anvi'o programs that profile BAM files, anvi-script-get-coverage-from-bam and anvi-profile-blitz.
|
|
🧀
bam-file |
|
🍕
single-profile-db |
|
🧠 |
🔥 anvi-profile-blitz. FAST profiling of BAM files to get gene-, contig-, or genome-level coverage and detection stats. Unlike anvi-profile, which is another anvi'o program that can profile BAM files, this program is designed to be very quick and only report long-format files for various read recruitment statistics per item. Plase also see the program anvi-script-get-coverage-from-bam for recovery of data from BAM files without an anvi'o contigs database.
|
|
🧀
bam-file |
|
🍕
bam-stats-txt |
|
🧠 |
| 🔥 anvi-reaction-network. This program generates a metabolic reaction network in an anvi'o contigs or pan database. |
|
🧀
contigs-db |
|
🍕
reaction-network |
|
🧠 |
| 🔥 anvi-refine. Start an anvi'o interactive interactive to manually curate or refine a genome, whether it is a metagenome-assembled, single-cell, or an isolate genome. |
|
🧀
profile-db |
|
🍕
bin |
|
🧠 |
| 🔥 anvi-rename-bins. Rename all bins in a given collection (so they have pretty names). |
|
🧀
collection |
|
🍕
collection |
|
🧠 |
| 🔥 anvi-reorient-genomes. Reorient circular genomes and scaffold fragmented genomes in a fasta-txt so their coordinates match a chosen reference genome using minimap2 alignment and seqkit manipulation.. |
|
🧀
fasta-txt |
|
🍕
fasta |
|
🧠 |
| 🔥 anvi-report-circularity. Predict contig circularity from paired-end read alignments in a given BAM file. This program samples insert sizes, looks for RF pairs spanning junctions, and reports per-contig circularity statistics.. |
|
🧀
bam-file |
|
🍕
contig-circularity-report-txt |
|
🧠 |
| 🔥 anvi-report-inversions. Reports inversions. |
|
🧀
bams-and-profiles-txt |
|
🍕
inversions-txt |
|
🧠 |
| 🔥 anvi-report-linkmers. Reports sequences stored in one or more BAM files that cover one of more specific nucleotide positions in a reference. |
|
🧀
bam-file |
|
🍕
linkmers-txt |
|
🧠 |
| 🔥 anvi-run-cazymes. Run dbCAN CAZymes on contigs-db. |
|
🧀
contigs-db |
|
🍕
functions |
|
🧠 |
| 🔥 anvi-run-hmms. This program deals with populating tables that store HMM hits in an anvi'o contigs database. |
|
🧀
contigs-db |
|
🍕
hmm-hits |
|
🧠 |
| 🔥 anvi-run-interacdome. Run InteracDome on a contigs database. |
|
🧀
contigs-db |
|
🍕
binding-frequencies-txt |
|
🧠 |
| 🔥 anvi-run-kegg-kofams. Run KOfam HMMs on an anvi'o contigs database. |
|
🧀
contigs-db |
|
🍕
kegg-functions |
|
🧠 |
| 🔥 anvi-run-ncbi-cogs. This program runs NCBI's COGs to associate genes in an anvi'o contigs database with functions. This program can also run NCBI's COGs to annotate an amino acid sequence with function. COGs database was been designed as an attempt to classify proteins from completely sequenced genomes on the basis of the orthology concept.. |
|
🧀
cogs-data |
|
🍕
functions |
🧠  |
| 🔥 anvi-run-pfams. Run Pfam on Contigs Database. |
|
🧀
contigs-db |
|
🍕
functions |
|
🧠 |
🔥 anvi-run-scg-taxonomy. The purpose of this program is to affiliate single-copy core genes in an anvi'o contigs database with taxonomic names. A properly setup local SCG taxonomy database is required for this program to perform properly. After its successful run, anvi-estimate-scg-taxonomy will be useful to estimate taxonomy at genome-, collection-, or metagenome-level).
|
|
🧀
contigs-db |
|
🍕
scgs-taxonomy |
|
🧠 |
🔥 anvi-run-trna-taxonomy. The purpose of this program is to affiliate tRNA gene sequences in an anvi'o contigs database with taxonomic names. A properly setup local tRNA taxonomy database is required for this program to perform properly. After its successful run, anvi-estimate-trna-taxonomy will be useful to estimate taxonomy at genome-, collection-, or metagenome-level)..
|
|
🧀
contigs-db |
|
🍕
trna-taxonomy |
|
🧠 |
| 🔥 anvi-run-workflow. Execute, manage, parallelize, and troubleshoot entire 'omics workflows and chain together anvi'o and third party programs. |
|
🧀
workflow-config |
|
🍕
workflow |
|
🧠 |
| 🔥 anvi-scan-trnas. Identify and store tRNA genes in a contigs database. |
|
🧀
contigs-db |
|
🍕
hmm-hits |
|
🧠 |
| 🔥 anvi-search-functions. Search functions in an anvi'o contigs database or genomes storage. Basically, this program searches for one or more search terms you define in functional annotations of genes in an anvi'o contigs database, and generates multiple reports. The default report simply tells you which contigs contain genes with functions matching to serach terms you used, useful for viewing in the interface. You can also request a much more comprehensive report, which gives you anything you might need to know for each hit and serach term. |
|
🧀
contigs-db |
|
🍕
functions-txt |
|
🧠 |
| 🔥 anvi-search-palindromes. A program to find palindromes in sequences. |
|
🧀
dna-sequence |
|
🍕
palindromes-txt |
|
🧠 |
| 🔥 anvi-search-primers. You provide this program with FASTQ files for one or more samples AND one or more primer sequences, and it collects reads from FASTQ files that matches to your primers. This tool can be most powerful if you want to collect all short reads from one or more metagenomes that are downstream to a known sequence. Using the comprehensive output files you can analyze the diversity of seuqences visually, manually, or using established strategies such as oligotyping.. |
|
🧀
samples-txt |
|
🍕
short-reads-fasta |
|
🧠 |
| 🔥 anvi-search-sequence-motifs. A program to find one or more sequence motifs in contig or gene sequences, and store their frequencies. |
|
🧀
profile-db |
|
🍕
misc-data-items |
|
🧠 |
| 🔥 anvi-self-test. A program for anvi'o to test itself. |
| 🧀 n/a |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-setup-cazymes. Download and setup CAZyme data from the dbCAN3. |
| 🧀 n/a |
|
🍕
cazyme-data |
|
🧠 |
| 🔥 anvi-setup-interacdome. Setup InteracDome data. |
| 🧀 n/a |
|
🍕
interacdome-data |
|
🧠 |
| 🔥 anvi-setup-kegg-data. Download and setup various databases from KEGG. |
| 🧀 n/a |
|
🍕
kegg-data |
|
🧠 |
| 🔥 anvi-setup-modelseed-database. This program downloads and sets up the ModelSEED Biochemistry database.. |
|
🧀
functions |
|
🍕
reaction-ref-data |
|
🧠 |
| 🔥 anvi-setup-ncbi-cogs. Download and setup NCBI's Clusters of Orthologous Groups database. |
| 🧀 n/a |
|
🍕
cogs-data |
|
🧠 |
| 🔥 anvi-setup-pdb-database. Setup or update an offline database of representative PDB structures clustered at 95%. |
| 🧀 n/a |
|
🍕
pdb-db |
|
🧠 |
| 🔥 anvi-setup-pfams. Download and setup Pfam data from the EBI. |
| 🧀 n/a |
|
🍕
pfams-data |
|
🧠 |
🔥 anvi-setup-scg-taxonomy. The purpose of this program is to download necessary information from GTDB (https://gtdb.ecogenomic.org/), and set it up in such a way that your anvi'o installation is able to assign taxonomy to single-copy core genes using anvi-run-scg-taxonomy and estimate taxonomy for genomes or metagenomes using anvi-estimate-scg-taxonomy).
|
| 🧀 n/a |
|
🍕
scgs-taxonomy-db |
|
🧠 |
🔥 anvi-setup-trna-taxonomy. The purpose of this program is to setup necessary databases for tRNA genes collected from GTDB (https://gtdb.ecogenomic.org/), genomes in your local anvi'o installation so taxonomy information for a given set of tRNA sequences can be identified using anvi-run-trna-taxonomy and made sense of via anvi-estimate-trna-taxonomy).
|
| 🧀 n/a |
|
🍕
trna-taxonomy-db |
|
🧠 |
| 🔥 anvi-setup-user-modules. Set up user-defined metabolic pathways into an anvi'o-compatible database. |
|
🧀
user-modules-data |
|
🍕
modules-db |
|
🧠 |
| 🔥 anvi-show-collections-and-bins. A script to display collections stored in an anvi'o profile or pan database. |
|
🧀
pan-db |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-show-misc-data. Show all misc data keys in all misc data tables. |
|
🧀
pan-db |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-split. Split an anvi'o pan or profile database into smaller, self-contained projects. Black magic.. |
|
🧀
profile-db |
|
🍕
split-bins |
|
🧠 |
| 🔥 anvi-summarize. Summarizer for anvi'o pan or profile db's. Essentially, this program takes a collection id along with either a profile database and a contigs database or a pan database and a genomes storage and generates a static HTML output for what is described in a given collection. The output directory will contain almost everything any downstream analysis may need, and can be displayed using a browser without the need for an anvi'o installation. For this reason alone, reporting summary outputs as supplementary data with publications is a great idea for transparency and reproducibility. |
|
🧀
profile-db |
|
🍕
summary |
|
🧠 |
| 🔥 anvi-summarize-blitz. FAST summary of many anvi'o single profile databases (without having to use the program anvi-merge).. |
|
🧀
single-profile-db |
|
🍕
quick-summary |
|
🧠 |
| 🔥 anvi-tabulate-trnaseq. A program to write standardized tab-delimited files of tRNA-seq seed coverage and modification results. |
|
🧀
trnaseq-contigs-db |
|
🍕
trnaseq-seed-txt |
| 🧠 |
| 🔥 anvi-trnaseq. A program to process reads from a tRNA-seq dataset to generate an anvi'o tRNA-seq database. |
|
🧀
trnaseq-fasta |
|
🍕
trnaseq-db |
|
🧠 |
| 🔥 anvi-update-db-description. Update the description in an anvi'o database. |
|
🧀
pan-db |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-update-structure-database. Add or re-run genes from an already existing structure database. All settings used to generate your database will be used in this program. |
|
🧀
contigs-db |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-script-add-default-collection. A script to add a 'DEFAULT' collection in an anvi'o pan or profile database with either (1) a single bin that describes all items available in the profile database, or (2) as many bins as there are items in the profile database wher every item has its own bin. The former is the default behavior that will be useful in most instances where you need to use this script. The latter is most useful if you are Florian and/or have something very specific in mind.. |
|
🧀
pan-db |
|
🍕
collection |
|
🧠 |
| 🔥 anvi-script-as-markdown. Markdownizides TAB-delmited data with headers in terminal.. |
| 🧀 n/a |
|
🍕
markdown-txt |
|
🧠 |
| 🔥 anvi-script-augustus-output-to-external-gene-calls. Takes in gene calls by AUGUSTUS v3.3.3, generates an anvi'o external gene calls file. It may work well with other versions of AUGUSTUS, too. It is just no one has tested the script with different versions of the program. |
|
🧀
augustus-gene-calls |
|
🍕
external-gene-calls |
|
🧠 |
| 🔥 anvi-script-checkm-tree-to-interactive. A helper script to convert CheckM trees into anvio interactive with taxonomy information. |
|
🧀
phylogeny |
|
🍕
interactive |
|
🧠 |
| 🔥 anvi-script-compute-ani-for-fasta. Run ANI between contigs in a single FASTA file. |
|
🧀
fasta |
|
🍕
genome-similarity |
|
🧠 |
| 🔥 anvi-script-compute-bayesian-pan-core. Runs mOTUpan on your gene clusters to estimate whether they are core or accessory. |
|
🧀
pan-db |
|
🍕
bin |
|
🧠 |
| 🔥 anvi-script-estimate-metabolic-independence. Takes a genome as a contigs-db, and tells you whether it can be considered as an organism of high metabolic independence, or not. |
|
🧀
contigs-db |
|
🍕
metabolic-independence-score |
|
🧠 |
| 🔥 anvi-script-filter-fasta-by-blast. Filter FASTA file according to BLAST table (remove sequences with bad BLAST alignment). |
|
🧀
contigs-fasta |
|
🍕
contigs-fasta |
|
🧠 |
🔥 anvi-script-filter-hmm-hits-table. Filter weak HMM hits from a given contigs database using a domain hits table reported by anvi-run-hmms..
|
|
🧀
contigs-db |
|
🍕
hmm-hits |
|
🧠 |
| 🔥 anvi-script-find-misassemblies. This script report errors in long read assembly using read-recruitment information. The input file should be a BAM file of long reads mapped to an assembly made from these reads.. |
|
🧀
bam-file |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-script-fix-homopolymer-indels. Corrects homopolymer-region associated INDELs in a given genome based on a reference genome. The most effective use of this script is when the input genome is a genome reconstructed by minION long reads, and the reference genome is one that is of high-quality. Essentially, this script will BLAST the genome you wish to correct against the reference genome you provide, identify INDELs in the BLAST results that are exclusively associated with homopolymer regions, and will take the reference genome as a guide to correct the input sequences, and report a new FASTA file. You can use the output FASTA file that is fixed as the input FASTA file over and over again to see if you can eliminate all homopolymer-associated INDELs. |
|
🧀
fasta |
|
🍕
fasta |
|
🧠 |
🔥 anvi-script-gen-defense-finder-models-to-hmm-directory. This program generates an anvi'o compatible HMM directory to be used with anvi-run-hmms from the MDMParis Defense Finder Models..
|
|
🧀
hmm-file |
|
🍕
hmm-source |
|
🧠 |
| 🔥 anvi-script-gen-distribution-of-genes-in-a-bin. Quantify the detection of genes in genomes in metagenomes to identify the environmental core. This is a helper script for anvi'o metapangenomic workflow. |
|
🧀
contigs-db |
|
🍕
view-data |
|
🧠 |
| 🔥 anvi-script-gen-function-matrix-across-genomes. A program to generate reports for the distribution of functions across genomes. |
|
🧀
functions |
|
🍕
functional-enrichment-txt |
|
🧠 |
| 🔥 anvi-script-gen-functions-per-group-stats-output. Generate a TAB delimited file for the distribution of functions across groups of genomes/metagenomes. |
|
🧀
functions |
|
🍕
interactive |
|
🧠 |
| 🔥 anvi-script-gen-genomes-file. Generate an external genomes or internal genomes file. |
|
🧀
contigs-db |
|
🍕
external-genomes |
|
🧠 |
| 🔥 anvi-script-gen-hmm-hits-matrix-across-genomes. A simple script to generate a TAB-delimited file that reports the frequency of HMM hits for a given HMM source across contigs databases. |
|
🧀
external-genomes |
|
🍕
hmm-hits-across-genomes-txt |
|
🧠 |
| 🔥 anvi-script-gen-pseudo-paired-reads-from-fastq. A script that takes a FASTQ file that is not paired-end (i.e., R1 alone) and converts it into two FASTQ files that are paired-end (i.e., R1 and R2). This is a quick-and-dirty workaround that halves each read from the original FASTQ and puts one half in the FASTQ file for R1 and puts the reverse-complement of the second half in the FASTQ file for R2. If you've ended up here, things have clearly not gone very well for you, and Evan, who battled similar battles and ended up implementing this solution wholeheartedly sympathizes. |
|
🧀
short-reads-fasta |
|
🍕
paired-end-fastq |
|
🧠 |
| 🔥 anvi-script-gen-short-reads. Generate short reads from contigs. Useful to reconstruct mock data sets from already assembled contigs. |
|
🧀
configuration-ini |
|
🍕
short-reads-fasta |
|
🧠 |
| 🔥 anvi-script-gen-user-module-file. This script generates a user-defined module file from a tab-delimited file of enzymes and other input parameters.. |
|
🧀
enzymes-list-for-module |
|
🍕
user-modules-data |
|
🧠 |
🔥 anvi-script-get-coverage-from-bam. Get nucleotide-level, contig-level, or bin-level coverage values from a BAM file very rapidly. For other anvi'o programs that are designed to profile BAM files, see anvi-profile and anvi-profile-blitz.
|
|
🧀
bam-file |
|
🍕
coverages-txt |
|
🧠 |
| 🔥 anvi-script-get-hmm-hits-per-gene-call. A simple script to generate a TAB-delimited file gene caller IDs and their HMM hits for a given HMM source. |
|
🧀
contigs-db |
|
🍕
functions-txt |
|
🧠 |
🔥 anvi-script-hmm-to-hmm-directory. You give this program one or more HMM files from hmmbuild, and it generates an anvi'o compatible HMM directory to be used with anvi-run-hmms.
|
|
🧀
hmm-file |
|
🍕
hmm-source |
|
🧠 |
| 🔥 anvi-script-merge-collections. Generate an additional data file from multiple collections. |
|
🧀
contigs-db |
| 🍕 n/a |
|
🧠 |
| 🔥 anvi-script-permute-trnaseq-seeds. This script generates a FASTA file of tRNA-seq seeds with permuted nucleotides at positions of predicted modification-induced substitutions. The underlying nucleotide without modification is not always the most common base call. The resulting FASTA file can be queried against a database of tRNA genes to validate nucleotides at modified positions and find the most similar sequences.. |
|
🧀
contigs-db |
|
🍕
contigs-fasta |
| 🧠 |
🔥 anvi-script-pfam-accessions-to-hmms-directory. You give this program one or more PFAM accession ids, and it generates an anvi'o compatible HMM directory to be used with anvi-run-hmms.
|
|
🧀
pfam-accession |
|
🍕
hmm-source |
|
🧠 |
🔥 anvi-script-process-genbank. This script takes a GenBank file, and outputs a FASTA file, as well as two additional TAB-delimited output files for external gene calls and gene functions that can be used with the programs anvi-gen-contigs-database and anvi-import-functions.
|
|
🧀
genbank-file |
|
🍕
contigs-fasta |
🧠  |
🔥 anvi-script-process-genbank-metadata. This script takes the 'metadata' output of the program ncbi-genome-download (see https://github.com/kblin/ncbi-genome-download for details), and processes each GenBank file found in the metadata file to generate a FASTA file, as well as genes and functions files for each entry. Plus, it autmatically generates a FASTA TXT file descriptor for anvi'o snakemake workflows. So it is a multi-talented program like that.
|
| 🧀 n/a |
|
🍕
contigs-fasta |
|
🧠 |
🔥 anvi-script-reformat-bam. Reformat a BAM file to match the updated sequence names after running anvi-script-reformat-fasta. You will need this script to fix your BAM file if you run anvi-script-reformat-fasta on a FASTA file of sequences after you already used the previous version of the FASTA file for read recruitment..
|
|
🧀
bam-file |
|
🍕
bam-file |
🧠  |
| 🔥 anvi-script-reformat-fasta. Reformat FASTA file (remove contigs based on length, or based on a given list of deflines, and/or generate an output with simpler names). |
|
🧀
fasta |
|
🍕
contigs-fasta |
🧠  |
| 🔥 anvi-script-snvs-to-interactive. Take the output of anvi-gen-variability-profile, prepare an output for interactive interface. |
|
🧀
variability-profile-txt |
|
🍕
interactive |
|
🧠 |
| 🔥 anvi-script-transpose-matrix. Transpose a TAB-delimited file. |
|
🧀
view-data |
|
🍕
view-data |
|
🧠 |
| 🔥 anvi-script-variability-to-vcf. A script to convert SNV output obtained from anvi-gen-variability-profile to the standard VCF format. |
|
🧀
variability-profile-txt |
|
🍕
vcf |
|
🧠 |