anvi-draw-kegg-pathways
Table of Contents
Write KEGG pathway map files incorporating data sourced from anvi'o databases.
🔙 To the main page of anvi’o programs and artifacts.
Authors

Can consume
contigs-db ![]() external-genomes
external-genomes ![]() pan-db
pan-db ![]() genomes-storage-db
genomes-storage-db ![]() kegg-data
kegg-data ![]()
Can provide
Usage
anvi-draw-kegg-pathways draws kegg-pathway-map files incorporating data from anvi’o databases. The visualization of user data in the context of KEGG’s curated biochemical pathways can reveal patterns in metabolism.
Setup
There are hundreds of pathway maps, listed and categorized here. anvi-setup-kegg-data downloads, among other files, the maps with corresponding XML files that allow elements of the map to be modified. The following command sets up the database in a default anvi’o directory.
anvi-setup-kegg-data
Additional Python packages may be needed if you installed anvi’o v8.0-dev before this program’s package requirements were included. These can be installed with the following command.
pip install biopython reportlab pymupdf
The program can be tested with the following command.
anvi-self-test --suite kegg-mapping
Download newest available files
Alternatively, KEGG data can be set up not from a snapshot but by downloading the newest files available from KEGG using the -D flag. In the following command, a higher number of download threads than the default of 1 is provided by -T, which significantly speeds up downloading.
anvi-setup-kegg-data -D -T 5
Install in non-default location
To preserve KEGG data that you’ve already set up for whatever reason, the new snapshot or download can be placed in a non-default location using the option, --kegg-data-dir.
anvi-setup-kegg-data --kegg-data-dir path/to/other/directory
anvi-draw-kegg-pathways requires a --kegg-dir argument to seek KEGG data in a non-default location.
Pathway selection
By default, this program draws the maps that contain data of interest, e.g., KO gene sequence annotations in a contigs-db.
To draw all maps available in kegg-data, including those that don’t contain data of interest, use the flag, --draw-bare-maps.
The option, --pathway-numbers, limits the output to maps of interest. A single ID number can be provided, e.g., 00010 for Glycolysis / Gluconeogenesis, or multiple numbers can be listed, e.g., 00010 00020. Regular expressions can also be provided, e.g., 011.. 01[23].., where . represents any character: here the set of numbers given by 011.. corresponds to “global” maps and 01[23].. to “overview” maps.
The following command would draw all global maps and the glycolysis map, regardless of whether they contain any anvi’o data of interest (here, KO annotations from a contigs database).
anvi-draw-kegg-pathways --contigs-dbs contigs-db \ --ko \ --pathway-numbers 011.. 00010 \ --draw-bare-maps \ -o output_dir
Output
This program requires the path to a directory as an argument to -o or --output-dir. This must be a non-existent directory unless the flag, -W or --overwrite-output-destinations, is also used. Options are available to make it easier to browse through output files and anticipate their contents.
File names
By default, output file names contain the ID of each map, e.g., kos_00010.pdf for Glycolysis / Gluconeogenesis. The --name-files flag attaches a simplified version of the pathway name to the file name, e.g., kos_00010_Glycolysis_Gluconeogenesis.pdf.
File categorization
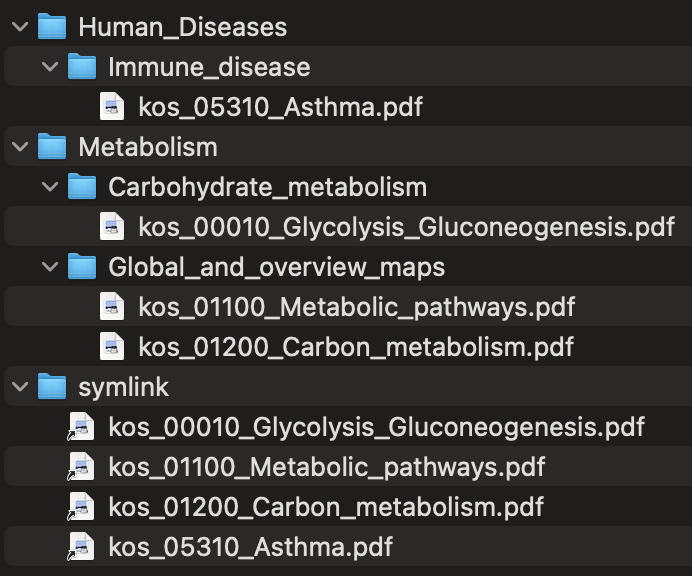
The --categorize-files flag categorizes output map files into a subdirectory structure based on the KEGG BRITE hierarchy of pathways. For example, a Glycolysis / Gluconeogenesis map would be placed in a directory named Metabolism/Carbohydrate_metabolism, as would be a Citrate cycle (TCA cycle) map, whereas an RNA polymerase map would be placed in a directory named Genetic_Information_Processing/Transcription. A subdirectory named symlink is also created with symbolic links to all of the categorized map files, allowing all of the files to be accessed from a single directory.
Here is a simple example of the output file structure produced with --name-files and --categorize-files in the course of anvi-self-test --suite kegg-mapping (with the -o option to save the temporary directories in the test from removal).
KO occurrence
Gene sequences in anvi’o databases can be annotated with KEGG Orthologs (KOs): see anvi-run-kegg-kofams. A KO indicates functional capabilities of the gene product. KO data from one or more contigs databases or a pan database can be mapped using the --ko flag, enabling investigation of the metabolic capabilities of individual organisms or multiple organisms, including community samples. Reactions associated with KOs are colored on the pathway maps.
Single contigs database
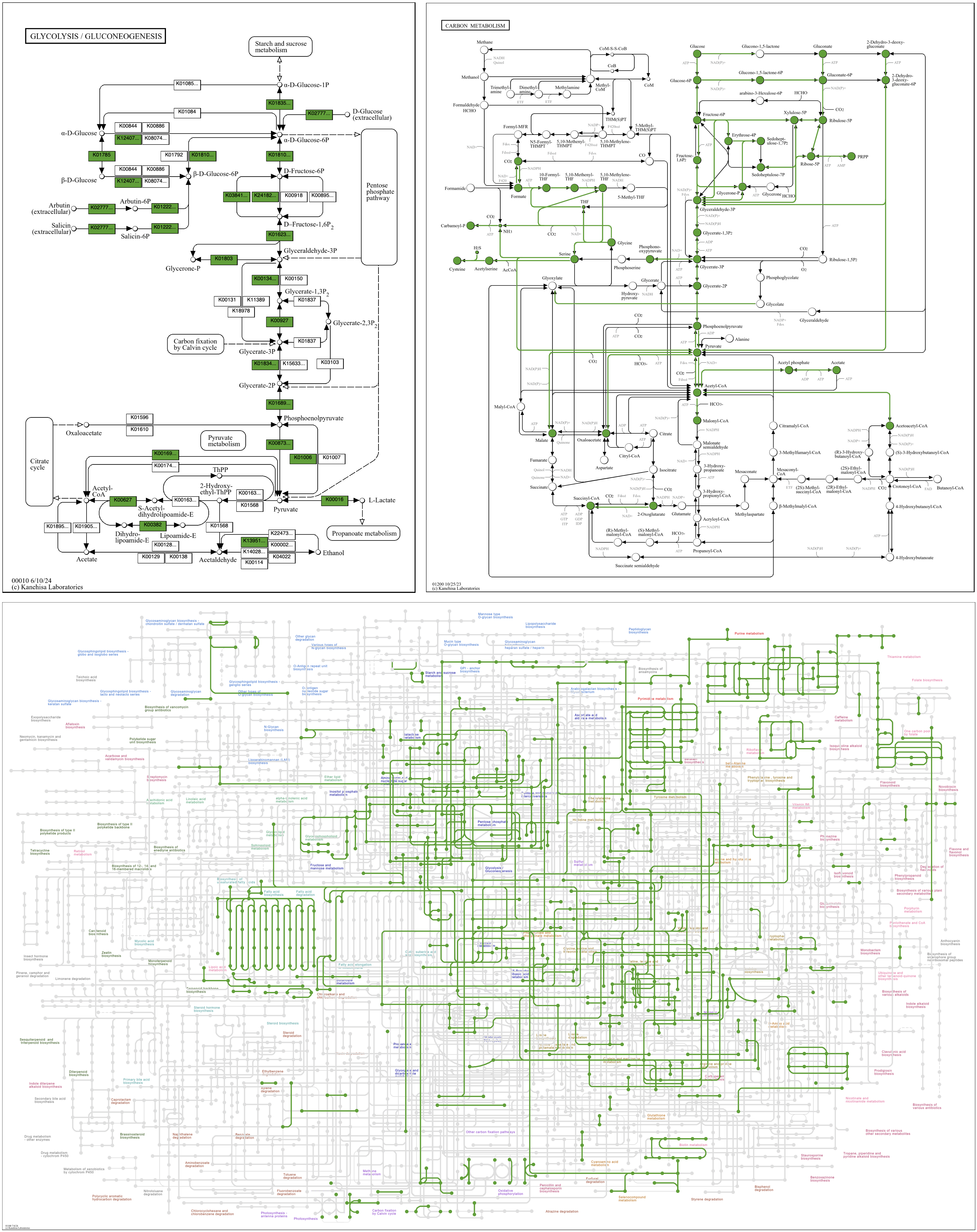
Here is the basic command to draw KO data from a single contigs-db.
anvi-draw-kegg-pathways --contigs-dbs contigs-db \ --ko \ -o output_dir
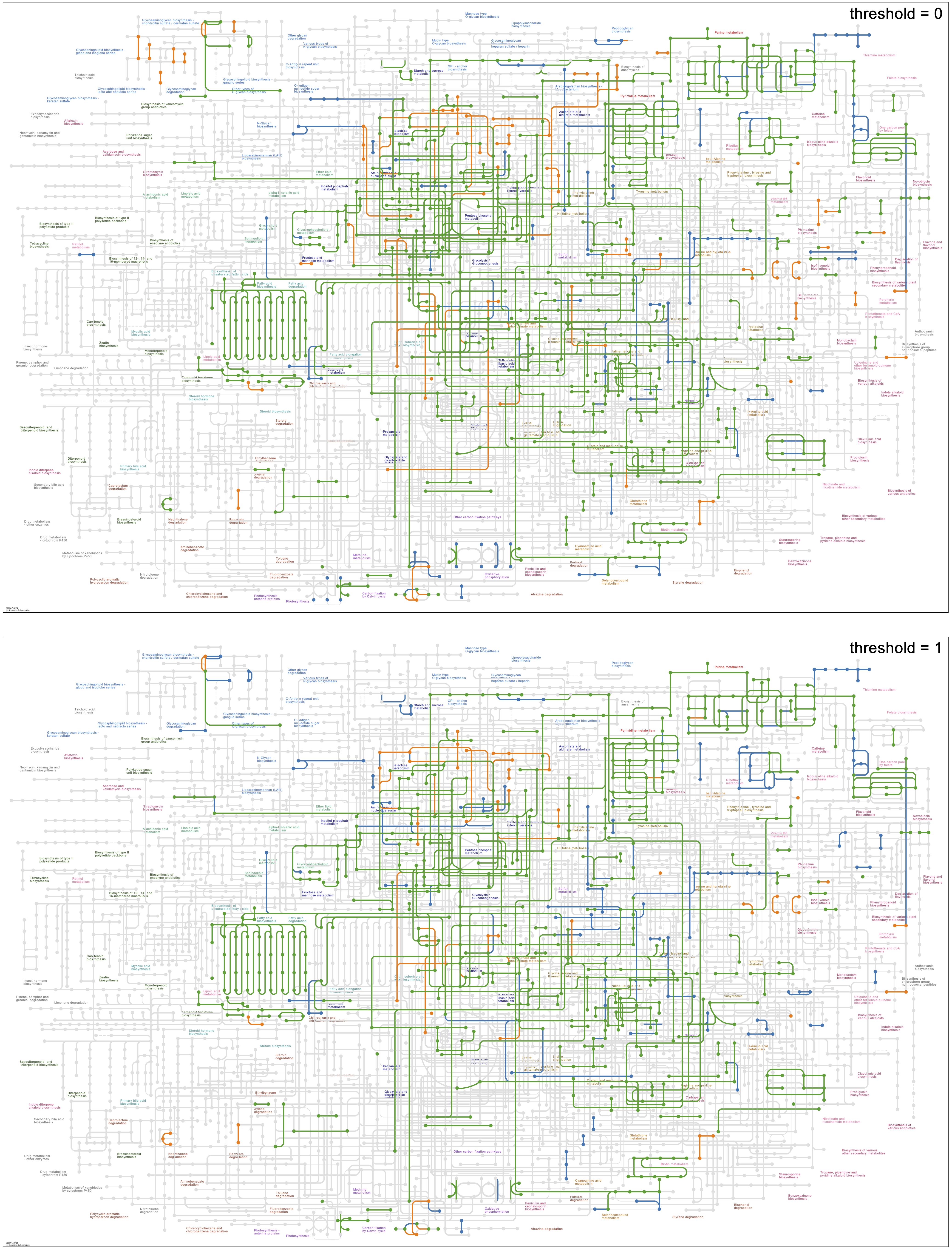
Here are three maps drawn with this command from a bacterial genomic contigs database. The map in the upper left, 00010 Glycolysis / Gluconeogenesis, is a “standard” map, in which boxes are associated with a reaction arrow and one or more KOs. The map in the upper right, 01200 Carbon metabolism, is a metabolic “overview” map. Overview maps have numerical IDs in the range 012XX and 013XX. Reaction arrows in overview maps are associated with one or more KOs and are colored and widened if represented by anvi’o KO data. The bottom map, 01100 Metabolic pathways, is a “global” metabolic map. Global maps have numerical IDs in the range 011XX. Reaction lines in global maps are associated with one or more KOs and colored if represented by anvi’o KO data. In all maps, circles are colored if the compound they represent is involved in reactions that are also colored. (Occasionally complete data linking reaction and compound graphics is missing from the KEGG reference files, preventing the reaction color from being imparted to the compound. One such error can be seen at the very top of the overview map of Carbon metabolism, where Glucono-1,5-lactone is white when it should be green.)
Set color
The default color can be changed with the --set-color option.
The argument can be a color hex code, e.g., "#FF0000" for red. It is necessary to enclose a color hex code argument in quotation marks, as # otherwise causes the rest of the command to be ignored as a comment.
anvi-draw-kegg-pathways --contigs-dbs contigs-db \ --ko \ --pathway-numbers 00010 \ --set-color “#2986cc” \ -o output_dir \
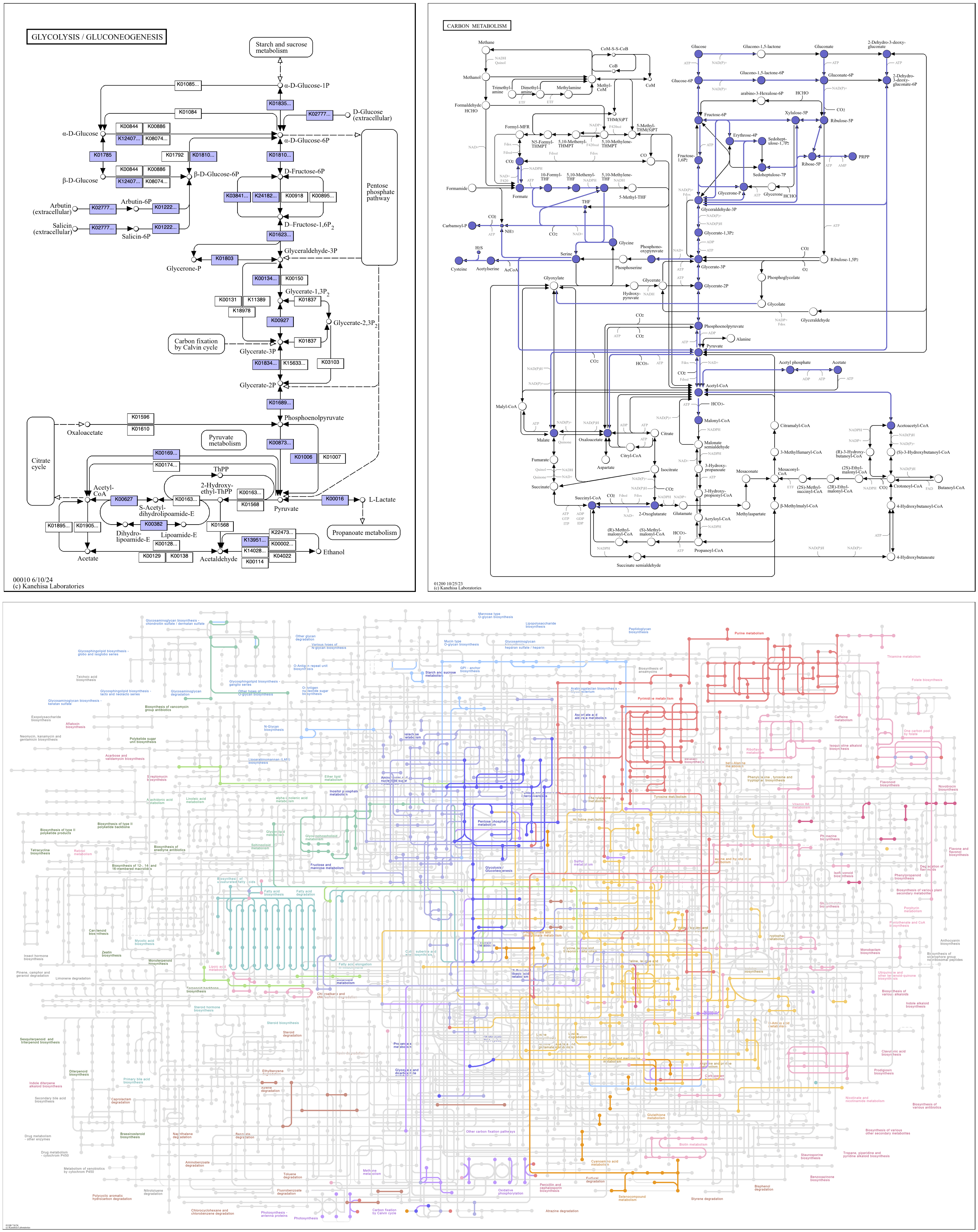
The argument can also be the string, original, for the original color scheme of the reference map. Global maps are especially colorful, with reactions varying in color across the map as a broad indication of function.
anvi-draw-kegg-pathways --contigs-dbs contigs-db \ --ko \ --pathway-numbers 00010 01100 01200 \ --set-color original \ -o output_dir
Compare multiple contigs databases
The KO content of multiple contigs databases can be compared. Database file paths can be provided directly on the command line or in an external-genomes text file.
anvi-draw-kegg-pathways --contigs-dbs contigs-db_1 contigs-db_2 … contigs-db_N \ --ko \ -o output_dir
anvi-draw-kegg-pathways --external-genomes external-genomes \ --ko \ -o output_dir
The images in this section show data from contigs databases of genomes from different strains of the same bacterial species.
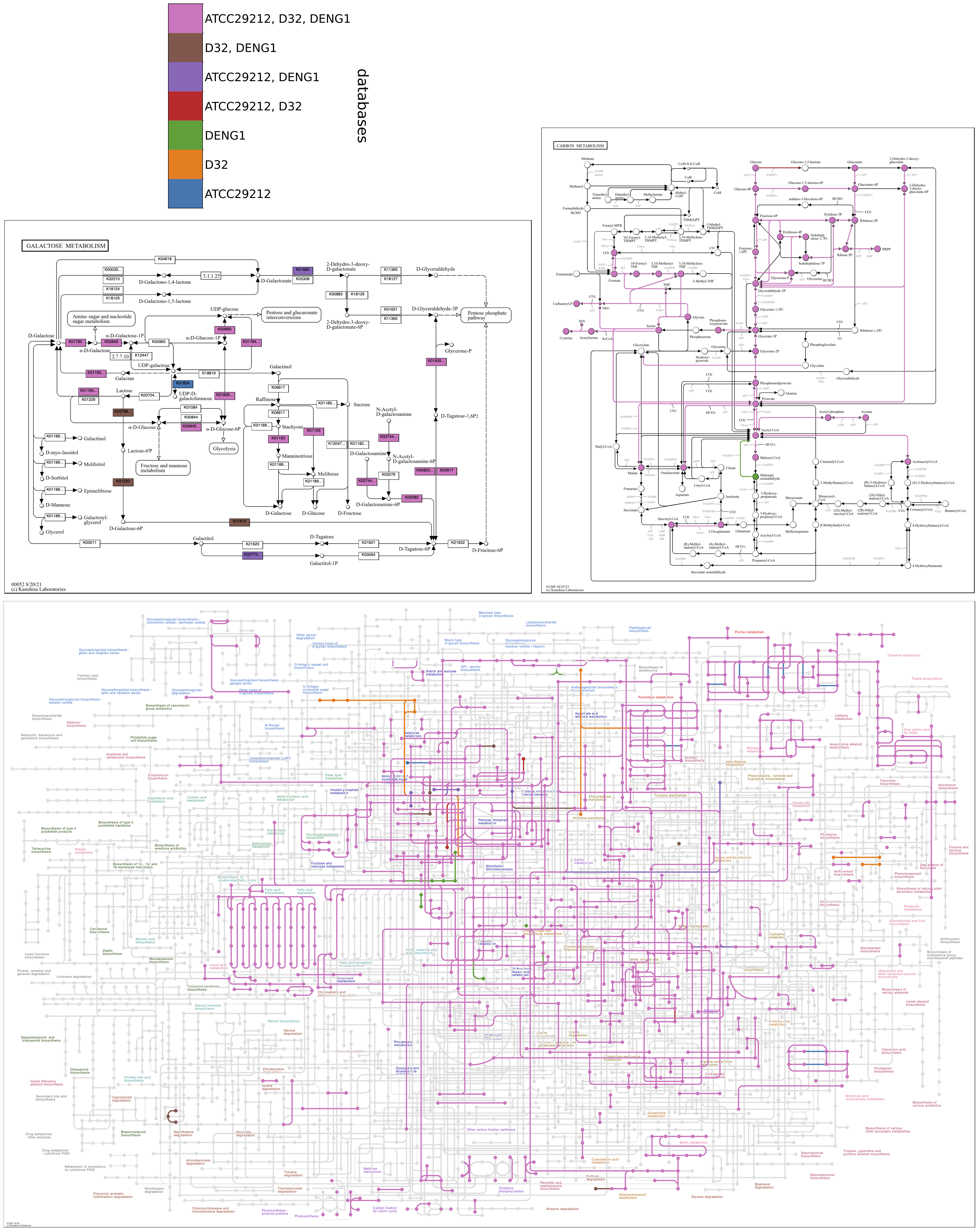
Color by database
When comparing a small number of contigs databases (realistically, two or three), reactions can be colored by their occurrence across databases, with each color representing a different database or combination of databases. A colorbar key is drawn in a separate file in the output directory, colorbar.pdf. Compound circles are imparted the color of the associated reaction found in the greatest number of databases.
Color by count
When comparing a larger number of contigs databases, it makes more sense to color reactions by the number of databases in which they occur using a sequential colormap rather than by database or combination of databases using a qualitative colormap. By default, coloring explicitly by database automatically applies to three or fewer databases, whereas coloring by database count applies to four or more databases. The user can override this default with the option, --colormap-scheme, which accepts the values by_source and by_count. For example, the user may have three databases but wish to color reactions by database count, and so would specify --colormap-scheme by_count.
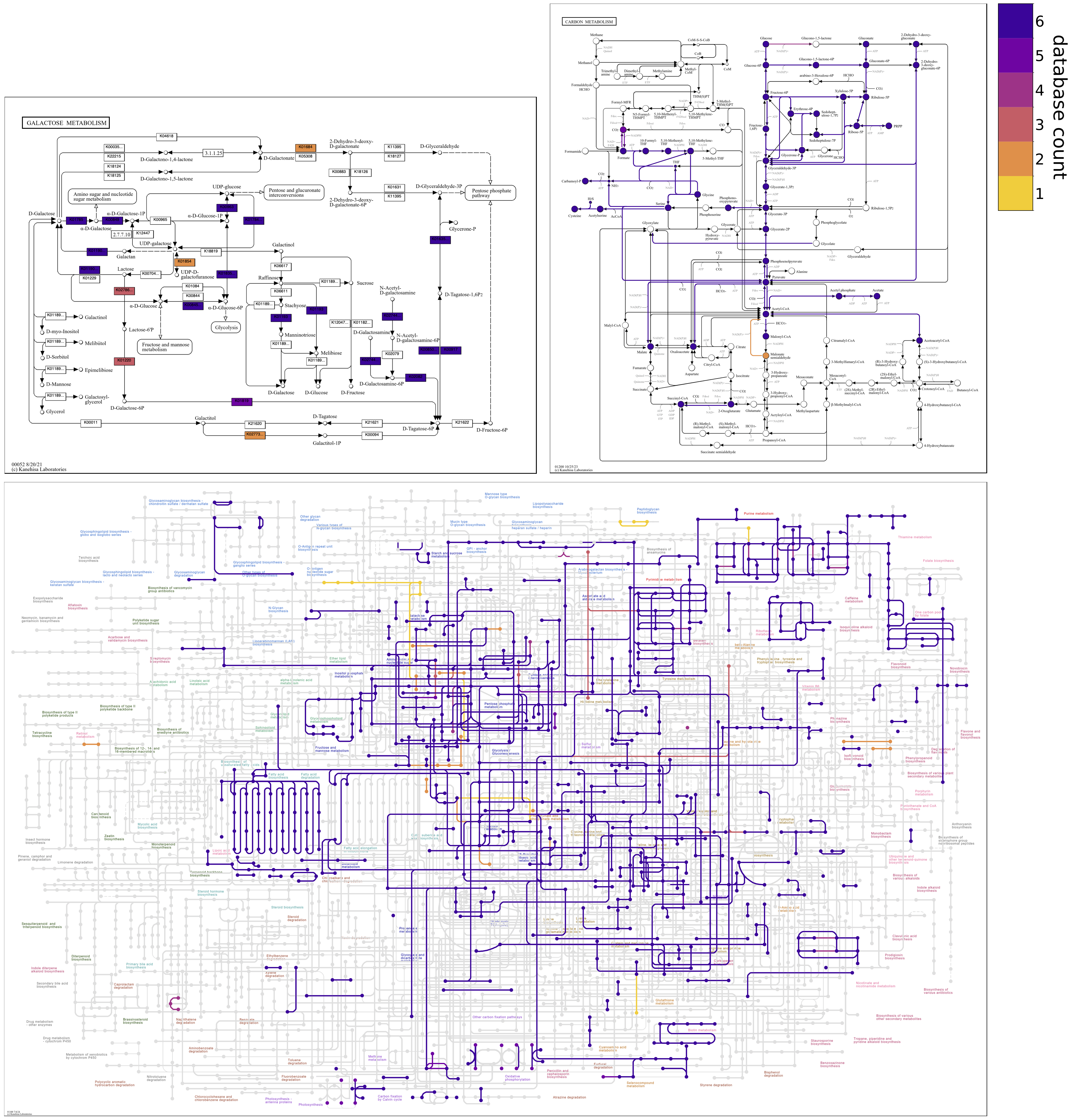
Reverse colormap
Changing the colormap can draw attention to different information on maps. When coloring by count, the default sequential colormap, plasma_r, goes from dark to light colors; reactions shared among all of the contigs databases are assigned the darkest color, and reactions unique to a single database are assigned the lightest color. The colormap can be reversed to accentuate unshared reactions in the darkest colors and shared reactions in the lightest colors. Reversing the default colormap is accomplished with the option, --colormap plasma 0.1 0.9. Note that Matplotlib colormap names differing by _r (here, plasma and plasma_r) have the same colors in reverse.
The second and third numerical --colormap values are not mandatory, but can be provided to trim a fraction of the colormap from each end to eliminate the lightest and darkest colors. The default coloring by database count with plasma_r uses limits of 0.1 0.9. Just changing the colormap (e.g., --colormap plasma) removes the limits (i.e., changes them to 0.0 1.0), so exactly reversing the default colormap requires that the same limits be specified.
The --reverse-overlay flag should also be used to reverse the default drawing order. This causes unshared reactions to be rendered above rather than below shared reactions, which is especially important in cluttered global maps.
anvi-draw-kegg-pathways --external-genomes external-genomes \ --ko \ --colormap plasma 0.1 0.9 \ --reverse-overlay \ -o output_dir
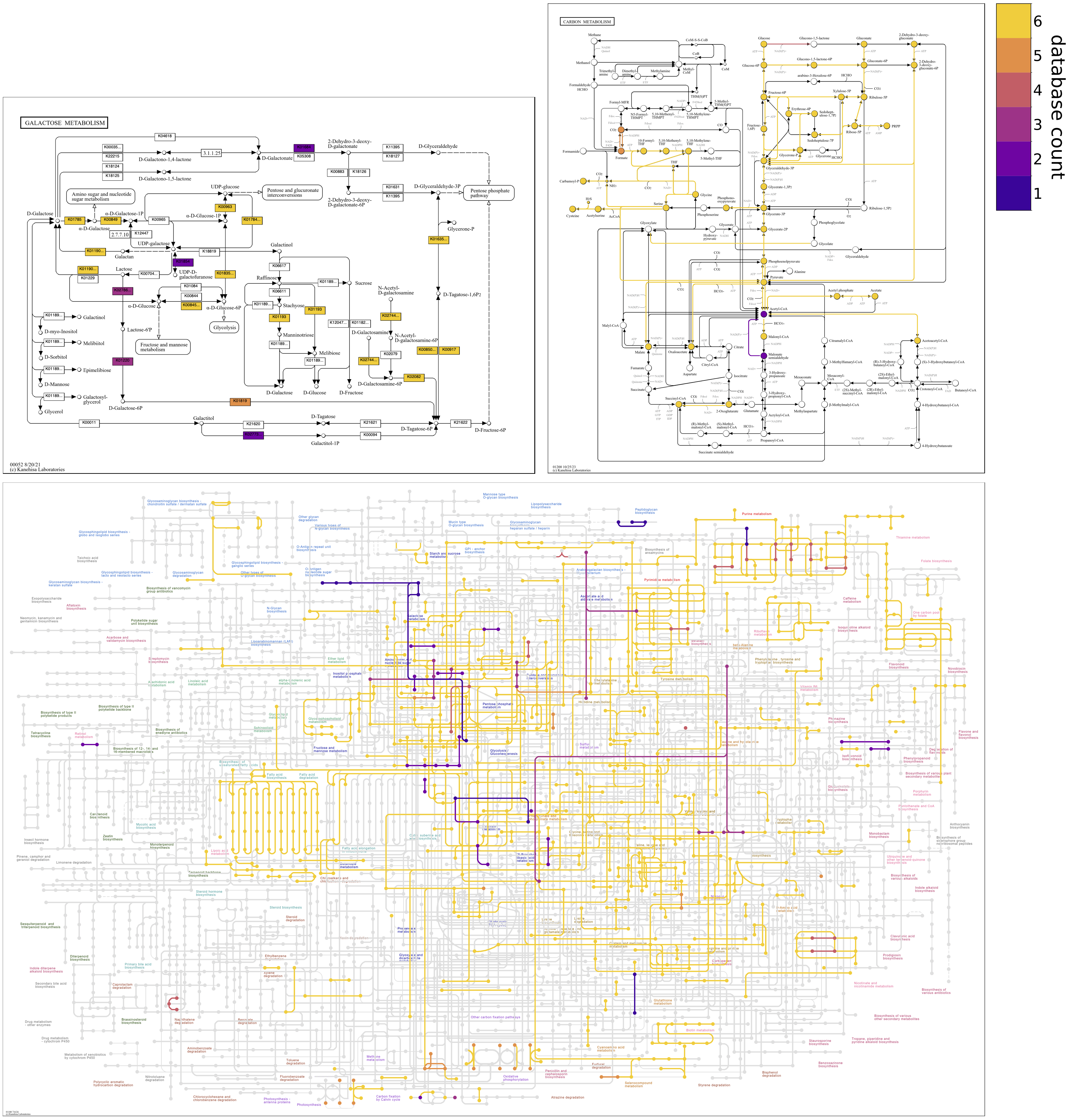
Show individual database maps
Coloring by count obviously masks the individual contigs databases that contain the different reactions. However, options are provided to enable investigation of the distribution of reactions across databases.
Standalone map files showing the presence/absence of reactions in all of the individual contigs databases can be drawn by using the option, --draw-individual-files, as a flag. Files can be drawn just for a subset of input databases by passing file arguments to the option, e.g., --draw-individual-files contigs_db_1 contigs_db_3. Individual map files for each database are stored in subdirectories of the output directory, with subdirectory names being the project names of databases.
To facilitate comparisons, maps for individual databases can also be drawn alongside the “unified” map containing information from all databases by using the option, --draw-grid, as a flag. Maps for just a subset of individual databases can be shown alongside the unified map in the grid file by passing file arguments to the option, e.g., --draw-grid contigs_db_2 contigs_db_3. Grid files are stored in a subdirectory of the output directory named grid.
The following command would draw individual map files plus grid files for all input contigs databases; a reverse colormap is used in unified maps to emphasize unshared reactions.
anvi-draw-kegg-pathways --external-genomes external-genomes \ --ko \ --draw-grid \ --draw-individual-files \ --colormap plasma 0.1 0.9 \ --reverse-overlay \ -o output_dir
The following map grid uncovers aspects of Galactose metabolism among the genomes of six Enterococcus faecalis strains. The unified map shows that some genomes may be missing steps from Lactose to D-Tagatose-6P in the tagatose-6-phosphate pathway, an alternative to the Leloir pathway for lactose catabolism found in some lactic acid bacteria. The individual maps show that strain ATCC29212 is missing three consecutive steps; OG1RF and V583 are missing two consecutive steps. When sequential reactions catalyzed by different enzymes are missing from a pathway in a closed genomic assembly while present in related genomes, it compellingly suggests that the genes and biological capacity are not in the genome rather than absent due to technical issues of orthology annotation.

Compare groups of contigs databases
A groups-txt file can be supplied to define groups of contigs databases. For example, databases representing genomes could be grouped by taxonomy, databases representing enrichment cultures under different conditions could be grouped by treatment, or databases representing marine metagenomic samples could be grouped by depth. The first column of groups-txt must contain the paths to the input contigs databases provided with --contigs-dbs. The second column headed group must contain group names, such as Pacific, Atlantic, and Arctic. Each database can only be assigned to one group.
A --group-threshold argument between 0 and 1 must also be provided to analyze groups. The group threshold is the proportion of databases in a group that must contain KOs defining a reaction on a map for the reaction to be associated with the group. A threshold of 0 means that ANY database in the group can contain the reaction for the reaction to be considered present in the group. A threshold of 0.75 means that at least 75% of databases in the group must contain the reaction for it to be present. A threshold of 1 means that ALL of the databases in the group must contain the reaction for it be present.
For example, set the threshold to 0.5. Reaction J on a map is defined by KO X and Reaction K is defined by KOs Y and Z. 90% of Pacific, 50% of Atlantic, and 10% of Arctic metagenomes contain KO X, so Reaction J would be colored to indicate that it is found in the Pacific and Atlantic. 0% of Pacific, 15% of Atlantic, and 40% of Arctic metagenomes contain KO Y or KO Z, so Reaction K would not be colored, being considered absent from the groups.
anvi-draw-kegg-pathways --external-genomes external-genomes \ --ko \ --groups-txt groups-txt \ --group-threshold 0.5 \ -o output_dir
As with comparisons of databases rather than groups, the default setting with two or three groups is to color reactions explicitly by the group or combination of groups in which they occur. With more databases, it makes more sense to color reactions by the number of databases in which they occur using a sequential colormap. The option --colormap-scheme can be used to override this behavior, e.g., to color reactions by group count rather than explicitly by membership using --colormap-scheme by_count.
The following example continues with Galactose metabolism, now including two groups of genomic contigs databases from Enterococcus faecalis and Enterococcus faecium. The program was run three times with different values of --group-threshold. The top map has a threshold of 0, so a reaction is included in the faecalis group if it has corresponding KOs in any of the six faecalis genomes, and included in the faecium group if it has KOs in any of the five faecium genomes. The middle map has a threshold of 0.5, so reactions in the faecalis group must be in at least four of six faecalis genomes, and reactions in the faecium group must be in at least three of five genomes. The bottom map has a threshold of 1. As in the map grid of faecalis genomes from above, the group maps show that some faecalis strains may be missing steps from Lactose to D-Tagatose-6P in the tagatose-6-phosphate pathway, though at least four of the strains have each step. In comparison, a greater proportion of the faecium strains have these steps, with all of them having the first two, as seen in the bottom map with a threshold of 1.
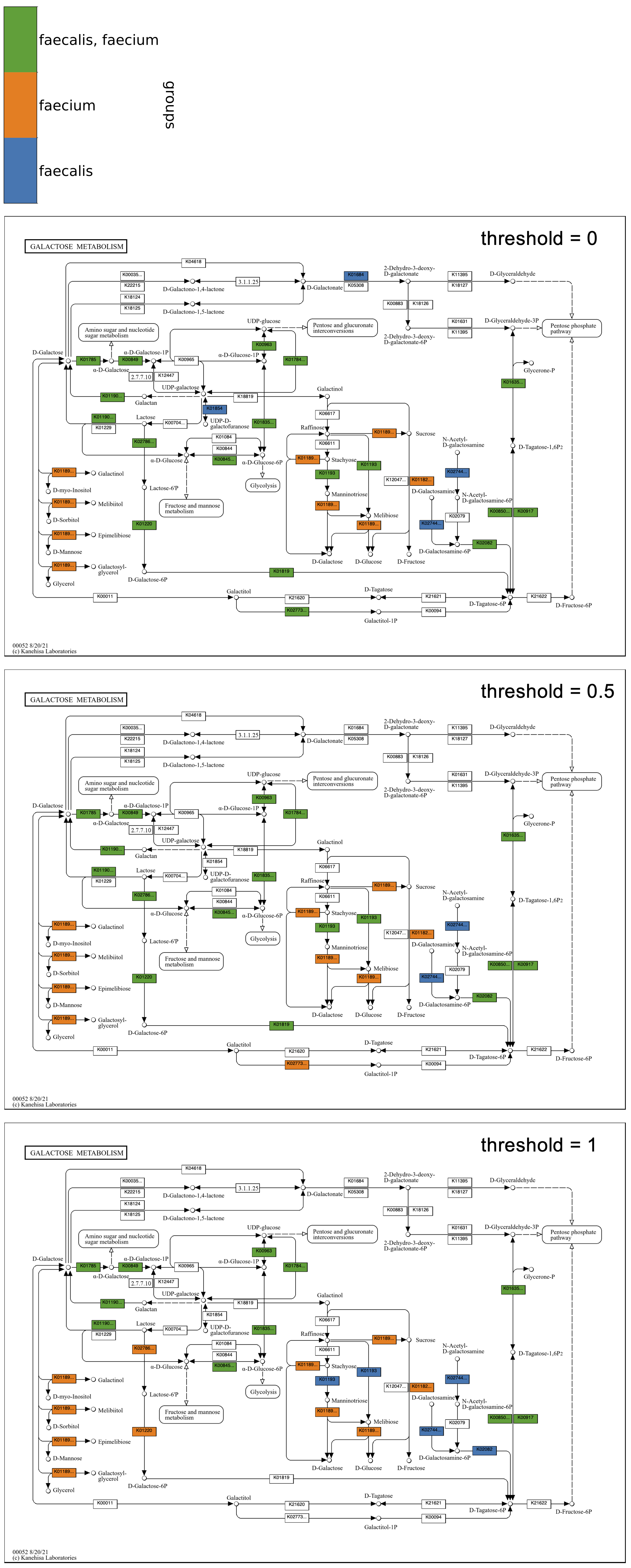
Below is the Global metabolism map using the same species groups with thresholds of 0 and 1. The map with a threshold of 0 shows how some metabolic pathways are present only in faecalis or faecium genomes, and the threshold of 1 shows how some of these unique pathways are conserved amongst all of the genomes of the species, such as the faecalis (blue) reactions in the upper right corner of the map involved in cofactor biosynthesis, and the faecium (orange) reactions in the upper center involved in sugar metabolism, including galactose, pentoses, and uronates.
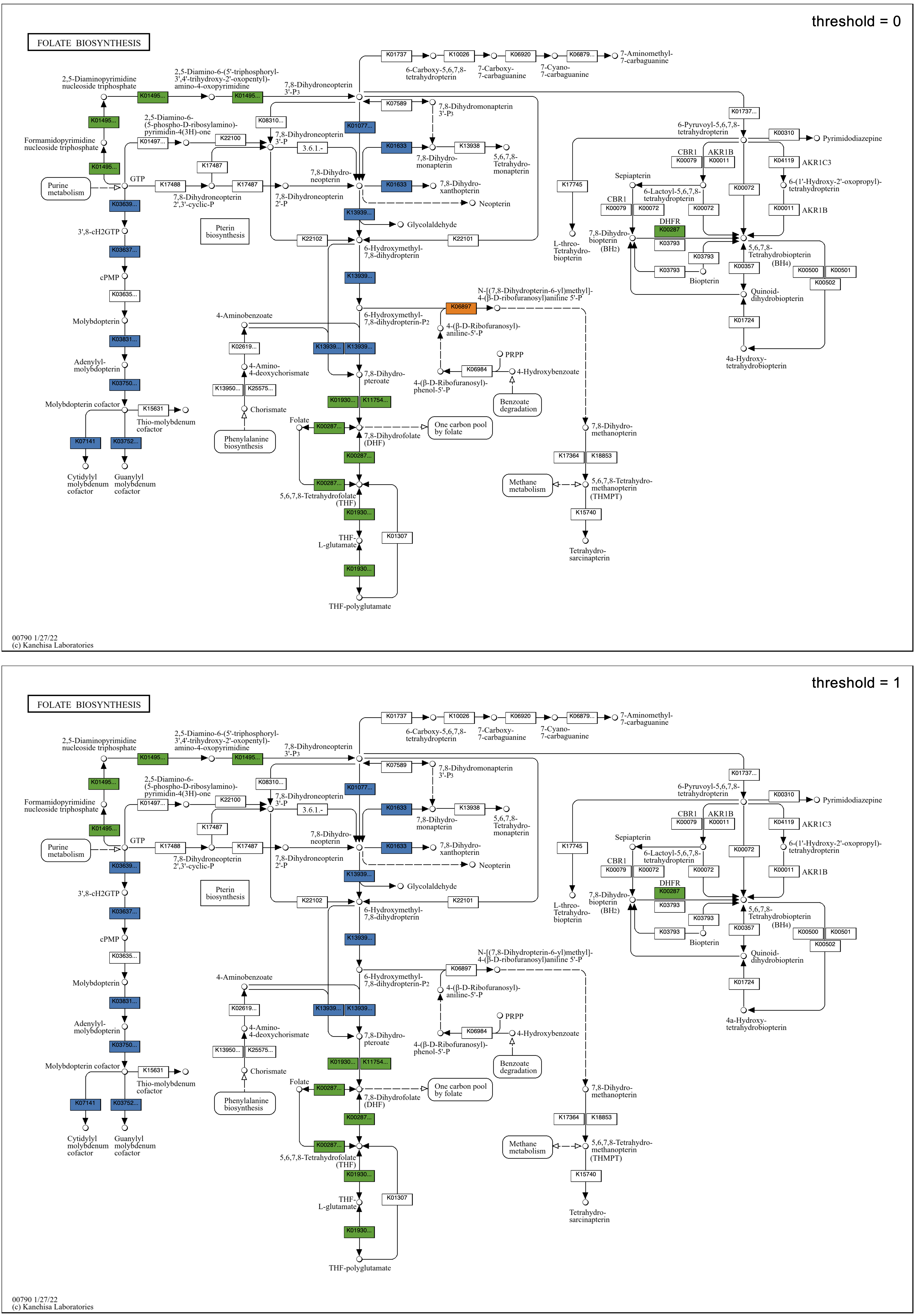
Exploring the Folate biosynthesis map given the observation regarding cofactors in the global map, all faecalis genomes have a full set of enzymes required for folate biosynthesis, whereas all faecium genomes appear to be missing a big chunk of the pathway in the center of the map, from 7,8-Dihydroneopterin 3'-3P to 7,8-Dihydropteroate. The implication of folate auxotropy in faecium is confirmed by Ramsey, Hartke, and Huycke in The Physiology and Metabolism of Enterococci: “Characteristically, E. faecium requires folate while E. faecalis does not.”
Show individual group maps
Maps can be drawn for individual groups as grids or separate files. These depict the occurrence of reactions across contigs databases in each group, identical to the maps that can be drawn to compare ungrouped databases.
Coloring options are available for individual group maps, --group-colormap and --group-reverse-overlay. Unlike drawing maps for ungrouped databases, drawing maps for individual groups always colors reactions by database count regardless of the number of databases in the group in order to facilitate the comparison of individual group maps using the same colormap scheme.
With the option, --draw-individual-files, individual group map files and a colorbar file are written to subdirectories of the output directory named after groups. With the option, --draw-grid, map grid files and colorbar files are written to the grid subdirectory of the output directory. There are colorbar files for each individual group in the grids: using our example Enterococcus dataset, there are files like colorbar_faecalis.pdf and colorbar_faecium.pdf.
A subset of groups can be considered, limiting the individual files drawn or the individual maps shown in grids to the specified groups, e.g., --draw-individual-files faecalis, --draw-grid faecium.
The following command would draw individual map files plus grid files for all groups. A reverse colormap is used in individual group maps to emphasize reactions that are not shared across databases of the group. The group threshold is set to 0, meaning a reaction is classified in a group if it occurs in any database, since the individual group maps reveal the number of databases in which the reaction occurs.
anvi-draw-kegg-pathways --external-genomes external-genomes \ --ko \ --groups-txt groups-txt \ --group-threshold 0 \ --draw-individual-files \ --draw-grid \ --group-colormap plasma 0.1 0.9 \ --group-reverse-overlay \ -o output_dir
Continuing with the comparison of Enterococcus species, the Folate biosynthesis map from above shows, on the left side, that all faecalis genomes have the pathway for molybdenum cofactor (MoCo) biosynthesis, unlike any faecium genomes. A molybdenum requirement in faecalis but not faecium is supported by the annotation of a molybdate transporter in all faecalis genomes and no faecium genomes, as seen in an ABC transporters map grid – in the map of “all” groups, it is the top transporter colored blue in the first column.

Pangenomic database
Pangenomes are treated similarly to multiple contigs databases. Rather than comparing the distribution of KOs across contigs databases, KOs assigned to pan-db gene clusters are compared across genomes. Here is the basic structure of the command.
anvi-draw-kegg-pathways -p pan-db \ -g genomes-storage-db \ -o output_dir \ --ko
The following maps were produced with the basic command structure for a pangenome of seven Enterococcus faecalis and five Enterococcus faecium genomes. A metagenome-assembled genome (MAG) has been added to the set of E. faecalis genomes from above.
Genomes defined in the pangenomic database can be grouped like contigs databases. The groups-txt file has the same format, but the items in the first column must now be the names of the genomes in the pangenome rather than contigs database files. The following command colors reactions by group, assigning a reaction to a group if the reaction is in at least 50% of the group’s genomes.
anvi-draw-kegg-pathways -p pan-db \ -g genomes-storage-db \ --ko \ --groups-txt groups-txt \ --group-threshold 0.5 \ -o output_dir
All of the options demonstrated in comparing contigs databases apply to pangenomes as well, including drawing map grids and changing the colormap to emphasize reactions unshared between genomes.
anvi-draw-kegg-pathways -p pan-db \ -g genomes-storage-db \ --ko \ --draw-grid \ --colormap plasma 0.1 0.9 \ --reverse-overlay \ -o output_dir
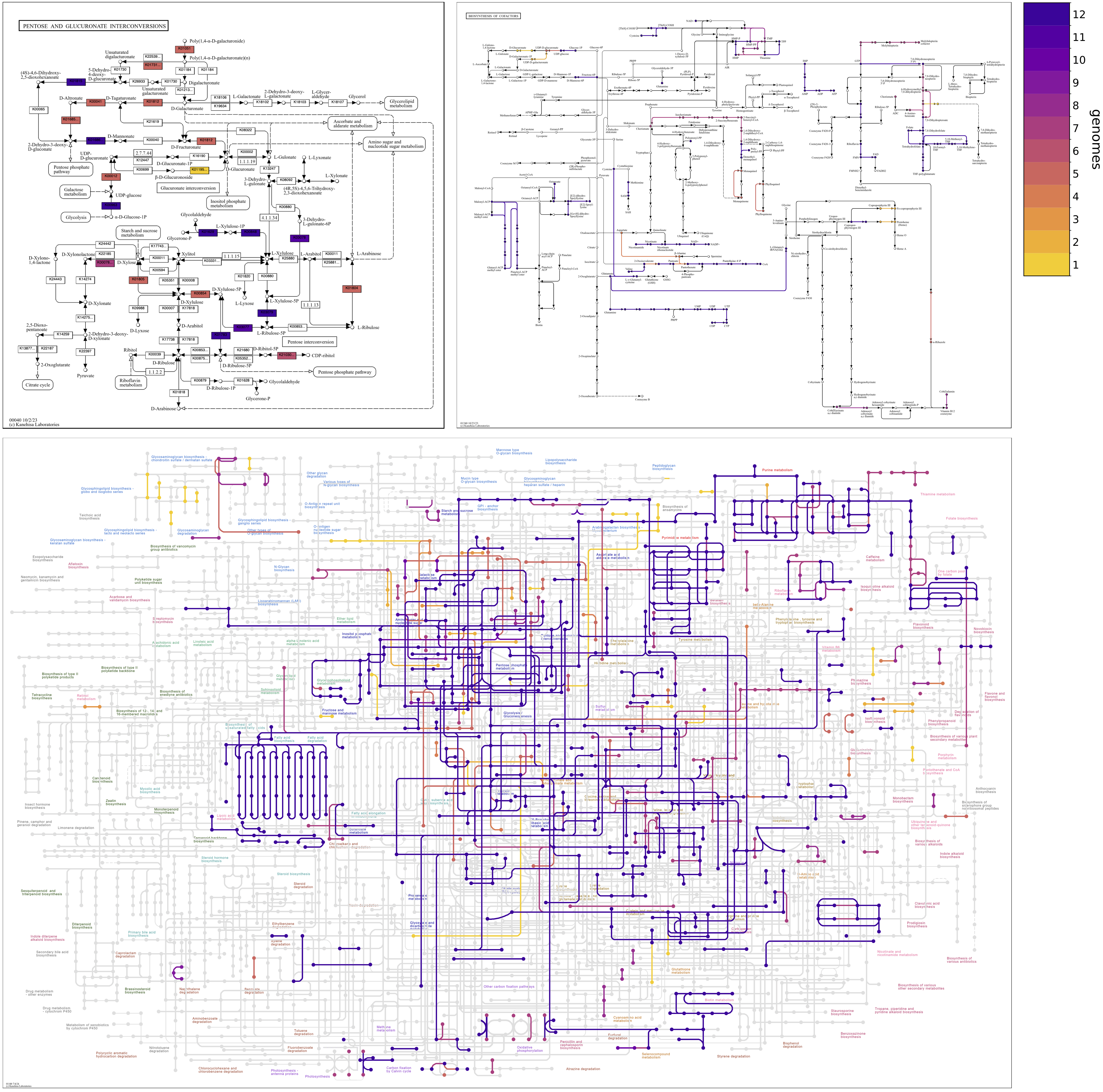
Continuing with the Enterococcus pangenome, the following map grid shows differences in Pentose and glucuronate interconversions between faecalis and faecium and between strains of each species. faecalis genomes are enriched in genes for xylose metabolism (towards the bottom of the map), and faecium genomes are enriched in enzymes for uronate metabolism (toward the top of the map). The faecalis MAG, SHARON, has genes for the catabolism of Xylose, via D-Xylulose and D-Xylulose-5P. These are also annotated in three of the faecalis isolate genomes, ATCC29212, DENG1, and V583, but not the other three, D32, OG1RF, and Symbioflor_1. Xylose utilization is known to be variable among faecalis strains.

Consensus KOs
The functional annotations of gene clusters as a whole are imputed to the genes in the cluster. The method of assigning consensus annotations to a cluster is parameterized by the options, --consensus-threshold and --discard-ties. By default, the consensus threshold is 0 and ties are not discarded. The consensus threshold sets the proportion of genes in the cluster that must have the same functional annotation to be assigned to the cluster as a whole, so a value of 0 means that the most abundant annotation is used regardless of its abundance. To discard ties means to ignore candidate consensus annotations that are equally abundant among genes. The default behavior randomly breaks ties. The consensus KO of a gene cluster becomes the KO annotation of all the genes in the cluster for the purposes of analysis.
Edit this file to update this information.
Additional Resources
Are you aware of resources that may help users better understand the utility of this program? Please feel free to edit this file on GitHub. If you are not sure how to do that, find the __resources__ tag in this file to see an example.