anvi-predict-metabolic-exchanges
Table of Contents
- Authors
- Requires
- Can use
- Provides
- Usage
- Prerequisites to using this program
- How to run this program
- Adjustable Parameters
- Setting some compound IDs as equivalent
- Using only one prediction method
- Skipping some Pathway Maps
- Including only specific Pathway Maps
- Changing the number of allowed gaps in the Pathway Map walks
- Using non-default data directories
- Adjusting number of threads and processes
- The program is running out of memory?
- Technical Details
- Cases we are looking for
- Using reaction networks to identify exchanged and unique compounds
- The “Reaction Network Subset” approach
- The “Pathway Map Walk” approach
- Additional Resources
Predicts metabolic exchanges between microbes based on the reaction network.
🔙 To the main page of anvi’o programs and artifacts.
Authors

Requires
Can use
reaction-network ![]() equivalent-compounds-txt
equivalent-compounds-txt ![]() external-genomes
external-genomes ![]() genome-pairs
genome-pairs ![]() reaction-ref-data
reaction-ref-data ![]() kegg-data
kegg-data ![]()
Provides
metabolite-exchange-predictions ![]()
Usage
anvi-predict-metabolic-exchanges uses the metabolic capacity encoded in two genomes to predict which metabolites could be exchanged between two organisms, and which metabolites are unique to one of the organisms. The output files produced by the program are described in metabolite-exchange-predictions.
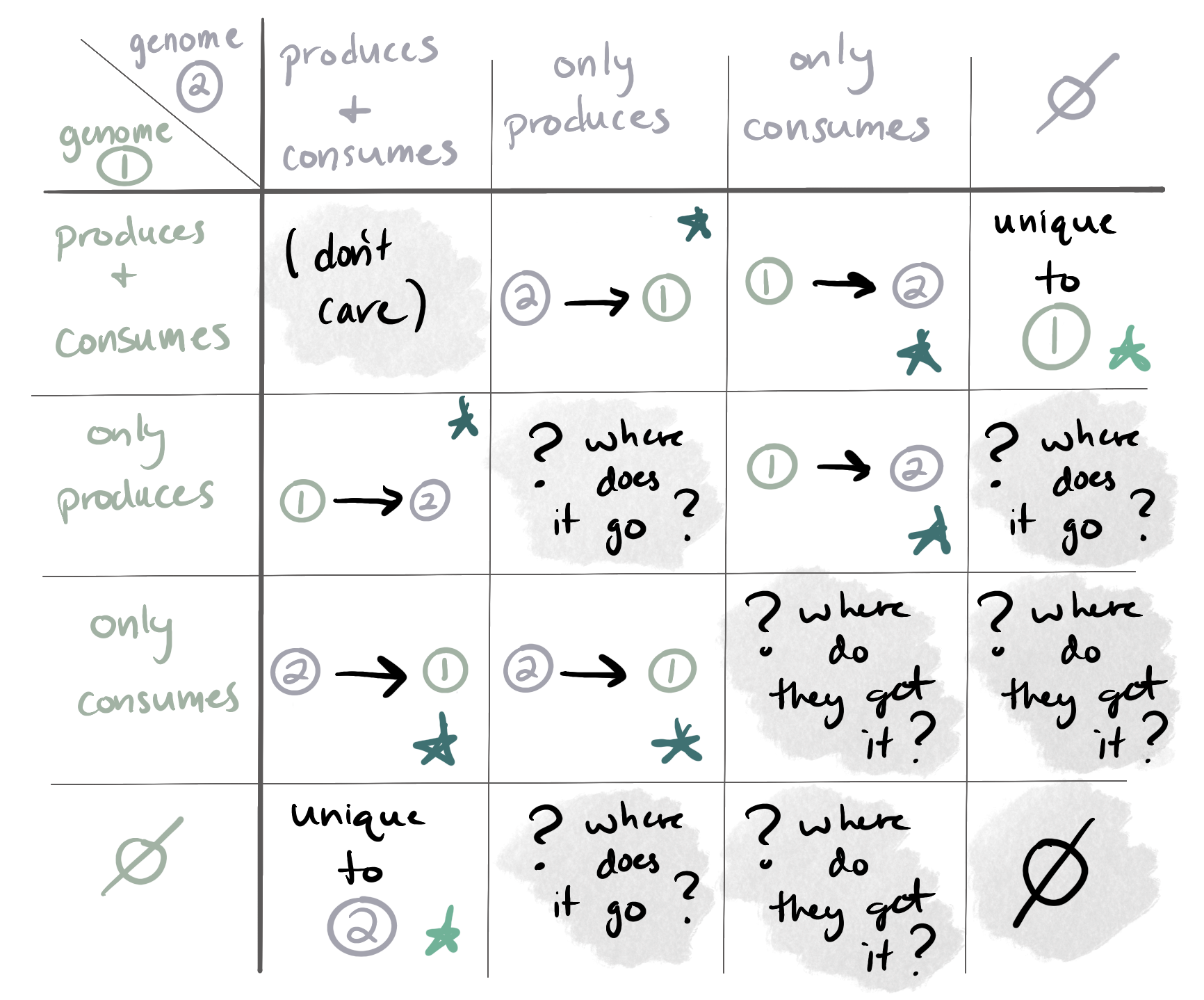
By leveraging the reaction-network infrastructure of anvi’o, the program examines the overlap between the metabolic reactions that each of the two organisms can catalyze and identifies which metabolic compounds fall into one of the following categories:
- can be produced by only one organism but consumed by the other (or both)
- can be consumed by only one organism but produced by the other (or both)
- can be produced and/or consumed by only one organism
It reports the metabolites in categories 1 and 2 as ‘potentially-exchanged compounds’, and the metabolites in category 3 as ‘unique’ compounds. Here is a table of what it looks for (everything marked with a star is reported by the program, and everything with a gray background is ignored):
Briefly, the program works by checking for these conditions for every metabolite in the genomes’ (merged) reaction networks. It does so in two orthogonal ways:
- Walking over KEGG Pathway Maps. For each compound that is part of a KEGG Pathway Map, we examine the chains of reactions that produce or consume the compound within the Pathway Map for each organism. This not only allows us to identify the aforementioned situations of potentially-exchanged or unique compounds, but also allows us to compute evidence for a given prediction (such as length and overlap of reaction chains) that can help you filter and interpret the output.
- Isolating metabolites in the merged reaction network. For each compound that is part of the genomes’ merged reaction network, we examine the subset of the network centered around it and determine whether it fits into one of the situations described above. This strategy currently doesn’t offer any supporting evidence for a given prediction (we only look at the potential ‘transfer point’ of the compound), but it does allow predictions for compounds that are not included in any KEGG Pathway Maps.
Either of these strategies can be skipped, in case you prefer to use only one method. If you want to know more, check the Technical Details section.
Prerequisites to using this program
This program uses the reaction-network stored in each contigs-db to search for potentially-exchanged or unique compounds. Since the reaction-network and this program both rely on the ModelSEED and KEGG databases (especially KEGG Pathway Maps and KOfam annotation profiles for KEGG Orthlogs), you need to have access to that data on your computer. You also need to have KEGG Ortholog (KO) annotations (kegg-functions) in your input genomes (which translates to having the functional annotation source ‘KOfam’ in the contigs-dbs).
Here are the steps you need to run before this program:
- anvi-setup-kegg-data to get data from KEGG onto your computer. This step only needs to be done once – if you’ve already ran this setup program in the past, you normally don’t have to do it again. When in doubt, skip this step and let anvi’o tell you if you are missing something.
- anvi-setup-modelseed-database to get data from ModelSEED onto your computer. Like the previous step, this only needs to be done once.
- anvi-run-kegg-kofams to annotate each of your contigs-dbs with KEGG Ortholog protein families.
- anvi-reaction-network to create a reaction network for each of of your contigs-dbs.
If you’ve done all that, you are good to go.
How to run this program
Running on a single pair of genomes
Provide both contigs databases for your genomes and a prefix for the output files:
anvi-predict-metabolic-exchanges -c1 contigs-db -c2 contigs-db \ -O ANY_PREFIX
The metabolite-exchange-predictions output file names will start using the prefix you provided.
MULTI-MODE: Running on multiple pairs of genomes
There are two ways for running this program in ‘multi-mode’ so that it predicts exchanges between multiple pairs of genomes. Both of them involve using an external-genomes file to point the program towards each contigs database. You will get one set of output files containing the predictions from all pairs.
If possible for your computer setup, we recommend increasing the number of threads using the --num-threads or -T parameter so that multiple pairs of genomes can be processed in parallel.
All-vs-all comparisons
If you provide only an external-genomes file, the program will determine all possible pairs of genomes and predict exchanges between all of them.
anvi-predict-metabolic-exchanges -e external-genomes \ -O ANY_PREFIX \ --num-threads 1
Specific pairwise comparisons
If you only want to predict exchanges between specific pairs of genomes, then you can enumerate those pairs in a genome-pairs file and provide that to the program (in addition to the external-genomes file).
anvi-predict-metabolic-exchanges -e external-genomes \ --genome-pairs-txt genome-pairs \ -O ANY_PREFIX \ --num-threads 1
Additional steps to run this program on internal genomes (bins)
If you have a collection of bin in a metagenome assembly, commonly described in an internal-genomes file, then you have to first run the program anvi-split to extract those bins into their own individual contigs databases. You will then need to run anvi-reaction-network on each bin’s individual contigs database, because running anvi-split does not split any reaction network made for the entire metagenome into subnetworks for each bin (at least, it did not do so at the time of writing this documentation).
Once you have a reaction-network in each bin’s contigs-db, you can create an external-genomes to use as input for anvi-predict-metabolic-exchanges as described above.
Adjustable Parameters
Setting some compound IDs as equivalent
The ModelSEED database sometimes has multiple compound ID numbers for what (we think) should be the same metabolite, at least for the purposes of predicting exchanges. A prime example of this is amino acids, which can have compounds where their chirality is specified (as in L-Lysine (cpd00039)) and where the chirality is generic (as in Lysine (cpd19182)). This is not so much a problem when we are using KEGG Pathway Maps to predict exchanges, but can lead to missing predictions when using the reaction network.
For amino acids, the program can automatically detect which compound IDs should be considered equivalent, and take that into account when looking for potentially-exchanged compounds. If you use the --use-equivalent-amino-acids flag, the program will search through the ModelSEED database for any conventional amino acids (plus Selenocysteine and Pyrrolysine) that have both an ‘L-‘ version and a chiral-unspecific version, and set those two compounds equivalent to each other.
anvi-predict-metabolic-exchanges -c1 contigs-db -c2 contigs-db \ -O ANY_PREFIX \ --use-equivalent-amino-acids
If you do this, you will get an output file listing the amino acid compounds that were deemed equivalent, so you can make sure you agree with them.
If you want to specify a custom set of equivalent compound IDs, you can instead provide an equivalent-compounds-txt file to the --custom-equivalent-compounds-file parameter:
anvi-predict-metabolic-exchanges -c1 contigs-db -c2 contigs-db \ -O ANY_PREFIX \ --custom-equivalent-compounds-file equivalent-compounds-txt
Using only one prediction method
If you want to skip the first prediction step of walking over KEGG Pathway Maps to find potential exchanges, use the --no-pathway-walk flag:
anvi-predict-metabolic-exchanges -c1 contigs-db -c2 contigs-db \ -O ANY_PREFIX \ --no-pathway-walk
If you want to skip the second prediction step of examining the local reaction network around each compound, use the --pathway-walk-only flag:
anvi-predict-metabolic-exchanges -c1 contigs-db -c2 contigs-db \ -O ANY_PREFIX \ --pathway-walk-only
It is hopefully understandable that these two flags are incompatible with each other.
Skipping some Pathway Maps
By default, this program will process all regular-type KEGG Pathway Maps (those with map IDs starting with prefix ‘00’) which have Reaction (‘RN’) type KGML files available in the KEGG data directory. If there are some Pathway Maps that you don’t want used for prediction (because they are irrelevant to your system, or you think they lead to misleading results, or they take too long to process, whatever), you can use the --exclude-pathway-maps option to provide a comma-separated list of Pathway Map ID numbers (just the numbers, no string prefix of any kind).
In the example below, we exclude two Pathway Maps: map 00195 (Photosynthesis) and map 00190 (Oxidative Phosphorylation). Both of these describe several enzyme complexes and do not explicitly contain any chemical reactions; hence, they do not have ‘RN’-type KGML files and would be skipped by the program anyway. The only effect of using the flag in this case would be that you don’t see any warnings on the terminal about those two Pathway Maps. However, if you use the flag to exclude a map that does have an ‘RN’-type KGML file, such as map 00470 (D-amino acid biosynthesis), the effect would be to remove any predictions (and accompanying evidence) that come from that Pathway Map.
anvi-predict-metabolic-exchanges -c1 contigs-db -c2 contigs-db \ -O ANY_PREFIX \ --exclude-pathway-maps 00195,00190
This option is only relevant if you allow the Pathway Map Walk prediction strategy (i.e., you didn’t use --no-pathway-walk).
Including only specific Pathway Maps
The opposite of --exclude-pathway-maps is the --include-pathway-maps option. With this flag, you can list the specific Pathway Maps that you want to predict exchanges from.
anvi-predict-metabolic-exchanges -c1 contigs-db -c2 contigs-db \ -O ANY_PREFIX \ --include-pathway-maps 00195,00190
Note that you may not get any predictions from the set of Pathway Maps you choose, so if you use --pathway-walk-only (to also skip the Reaction Network predictions), your output files could be completely empty.
Changing the number of allowed gaps in the Pathway Map walks
The --maximum-gaps parameter applies to the first prediction step of walking over KEGG Pathway Maps, and allows a certain number of missing enzyme annotations in the reaction chains. By default, we don’t allow any gaps, but if you think missing annotations in either genome might be throwing off your predictions, you can set this parameter to an integer greater than 0:
anvi-predict-metabolic-exchanges -c1 contigs-db -c2 contigs-db \ -O ANY_PREFIX \ --maximum-gaps 1
Changing this parameter will mostly affect the evidence attributed to a given prediction (i.e., length and overlap of reaction chains), but there could be some cases where increasing the gap number enables new predictions to be made.
Using non-default data directories
If you set up your KEGG or ModelSEED data in a custom directory, you can make sure this program knows where to find it by providing the paths:
anvi-predict-metabolic-exchanges -c1 contigs-db -c2 contigs-db \ -O ANY_PREFIX \ --kegg-data-dir /path/to/directory/KEGG \ --modelseed-data-dir /path/to/directory/MODELSEED
The data directories are relevant for loading the reaction-network in the contigs database, so it is best to use the same data directories that were utilized when running anvi-reaction-network.
Adjusting number of threads and processes
If you have a lot of computational resources, you can make this program work faster by parallelizing some of the tasks. For instance:
- when processing a single pair of genomes, multiple Pathway Map Walks can be done at once according to the number of threads set using the parameter
--num-threads - when processing multiple pairs of genomes (i.e., ‘MULTI-MODE’), multiple pairs of genomes can be processed at once according to the number of threads set using the
--num-threadsparameter. Each comparison, however, will internally utilize one thread, UNLESS… - since MULTI-MODE internally does multiple ‘single pair’ comparisons, you can increase the number of threads each comparison uses for Pathway Map walks by setting both the
--num-parallel-processesparameter and the--num-threadsparameter
Confused? Let’s see a few examples.
Multithreading ‘single’ mode
In this example, the program will predict exchanges between one pair of genomes, and it will be able to do Pathway Map walks for 4 Pathway Maps at once.
anvi-predict-metabolic-exchanges -c1 contigs-db -c2 contigs-db \ -O ANY_PREFIX \ --num-threads 4
Resource requirement: Your computer (or the job you submitted to a high-performance computing cluster) must have >4 threads available to run this example.
Why >4 and not >=4? I’m glad you asked. You see, the program itself has to run on one thread and it will start 4 child processes to work on the Pathway Map walks, bringing the total usage up to n=5 threads. Why not spawn n-1 child processes so that the total thread usage of the program is exactly equal to the --num-threads number? Well, that is a good idea. But in practice, the program (parent process) is not doing very much while waiting for its child processes to send their data back. And it seemed less confusing this way to begin with. If you think that extra thread will cause issues, now you know and you can set --num-threads a bit lower accordingly. And if you are upset about this, you know who to complain to. 😇
Multithreading multi-mode (multiple single-threaded comparisons at once)
In this example, the program will predict exchanges between all possible pairs of genomes in the provided external-genomes file, and it will be able to process 4 genome pairs at once. Each genome pair comparison is single-threaded in this case, meaning that there is no parallel processing of Pathway Map walks.
anvi-predict-metabolic-exchanges -e external-genomes \ -O ANY_PREFIX \ --num-threads 4
Resource requirement: Your computer (or the job you submitted to a high-performance computing cluster) must have >4 threads available to run this example.
Multiprocessing multi-mode (multiple multithreaded comparisons at once)
Finally, in this example, the program will process multiple pairs of genomes, but each genome pair comparison is multithreaded. Specifically, here we allow it to process 3 genome pairs in parallel, and each parallel process can in turn use 4 threads (so the Pathway Map Walk is multithreaded).
anvi-predict-metabolic-exchanges -e external-genomes \ -O ANY_PREFIX \ --num-threads 4 \ --num-parallel-processes 3
Resource requirement: Your computer (or the job you submitted to a high-performance computing cluster) must have >12 threads available to run this example.
Why >12 threads? Well, 4 * 3 = 12, plus there is one additional thread on which the main program is running while waiting for its child processes for a total usage of n=13 threads (confused? read the yellow box under the first example). If you are paying really close attention, you might realize that each child process needs to run on its own thread while spawning its own set of (grand)child processes. Good catch! We account for this by allowing each child process to create t-1 threads (where t is the --num-threads value) so that the total per-process footprint is equal to --num-threads. So in this example, technically only 3 Pathway Map walks are done at once for each genome pair comparison even though we set --num-threads to 4. At large scales, we cannot necessarily afford to ignore an extra thread per child process like we do for the parent process.
The program is running out of memory?
If this program starts consuming tons of memory and taking ages to run at the Pathway Map walk step, it’s likely stuck in an infinite cycle of reactions for one of the Pathway Maps. We have sanity checks to prevent this sort of thing, but we cannot guarantee that we’ve identified all problematic cases. The good thing is that these sort of issues are input-dependent, so you can avoid them by excluding problematic maps with the --exclude-pathway-maps flag (if you cannot figure out which maps are problematic from the regular program output, try re-running the program with --debug).
If you don’t want to exclude specific Maps entirely from your analysis, one workaround is to use the --max-reactions-for-pathway-map-walk parameter to truncate problematic walks early. Note that this upper bound on reaction chain length applies globally, so it’s probably best to use it only on problematic Pathway Maps:
anvi-predict-metabolic-exchanges -e external-genomes \ -O ANY_PREFIX \ --max-reactions 25 \ --include-pathway-maps 00061
Just keep in mind when you interpret the results that all reaction chain lengths in the evidence output will be artificially capped at the value you set, and that long chains could be technical artifacts from cycles rather than biologically relevant.
Technical Details
This section describes how the predictions of metabolic exchanges are done, for a given pair of genomes.
Cases we are looking for
You might recall the table of metabolite production vs consumption possibilities at the top of this documentation page. Here it is again so that you don’t have to scroll up:
To identify potentially-exchanged compounds, we look for all the cases marked by a dark green star; that is, compounds for which one of the genomes can either only produce or only consume the metabolite, such that it is possible that this genome is passing the metabolite to the other genome or receiving it from the other genome, respectively.
To identify unique compounds, we look for all the cases marked by a light green star in the table; that is, compounds for which production and/or consumption can only be done by one of the two genomes. We ignore all the other possibilities (gray background in the table) because the genomes either have no biological motivation to share the metabolite (that we can detect) or the situation raises questions that we simply cannot answer computationally.
How do we determine which organism is the ‘primary producer’ and which organism is the ‘primary consumer’ for potentially-exchanged compounds?
In some cases, more than one of the organisms can produce/consume a metabolite, but we have to pick just one of the two as the ‘primary’ producer or consumer of the compound. This is how we do that for a compound that is potentially-exchanged:
- if only one of the organisms can produce the compound, it is considered the ‘primary producer’ and the other organism is assigned by default to the role of ‘primary consumer’ (even if the first organism can also consume the compound)
- if only one of the organisms can consume the compound, it is considered the ‘primary consumer’ and the other organism is assigned by default to the role of ‘primary producer’ (even if the first organism can also produce the compound)
This role assignment is mostly relevant for interpreting the Pathway Walk evidence (described in a later section). The output of the program will describe all genomes that can ‘produce’ or ‘consume’ a compound when relevant, so you won’t be missing any information about who can do what.
Using reaction networks to identify exchanged and unique compounds
In order to start searching for the cases described above, we need to first identify the capacity for production or consumption of a metabolite by looking for the enzymes that can catalyze chemical reactions with the metabolite as a product or as a substrate, respectively. This is done using the reaction-networks generated by anvi-reaction-network. Each genome’s reaction-network matches the KEGG KOfam annotations of the genome to chemical reactions in the ModelSEED database, and includes all the enzymes/reactions/metabolites that are theoretically usable by the organism.
anvi-predict-metabolic-exchanges therefore starts by merging the two genome’s reaction networks together, to make one big network that tracks which reactions are catalyzable by each genome:
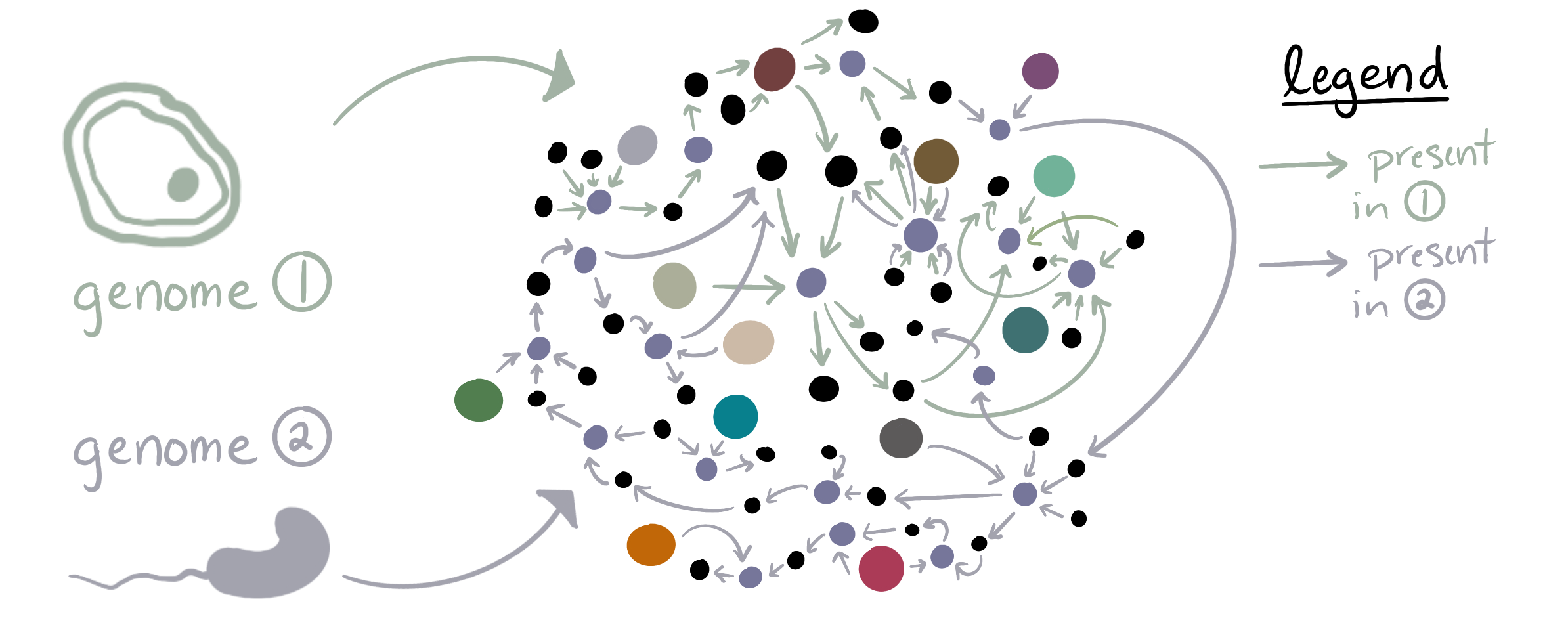
We can then iterate over every single metabolite within the merged network to identify whether it represents into one of our cases of interest from the table. Conceptually, this means looking at the production/consumption reactions surrounding the metabolite within the network and asking which genome(s) can produce/consume the compound. Here is an example in which the compound is considered ‘potentially-exchanged’ because only one genome (the purple one) can produce the compound, while both genomes have consumption reactions for it (purple and green arrows):
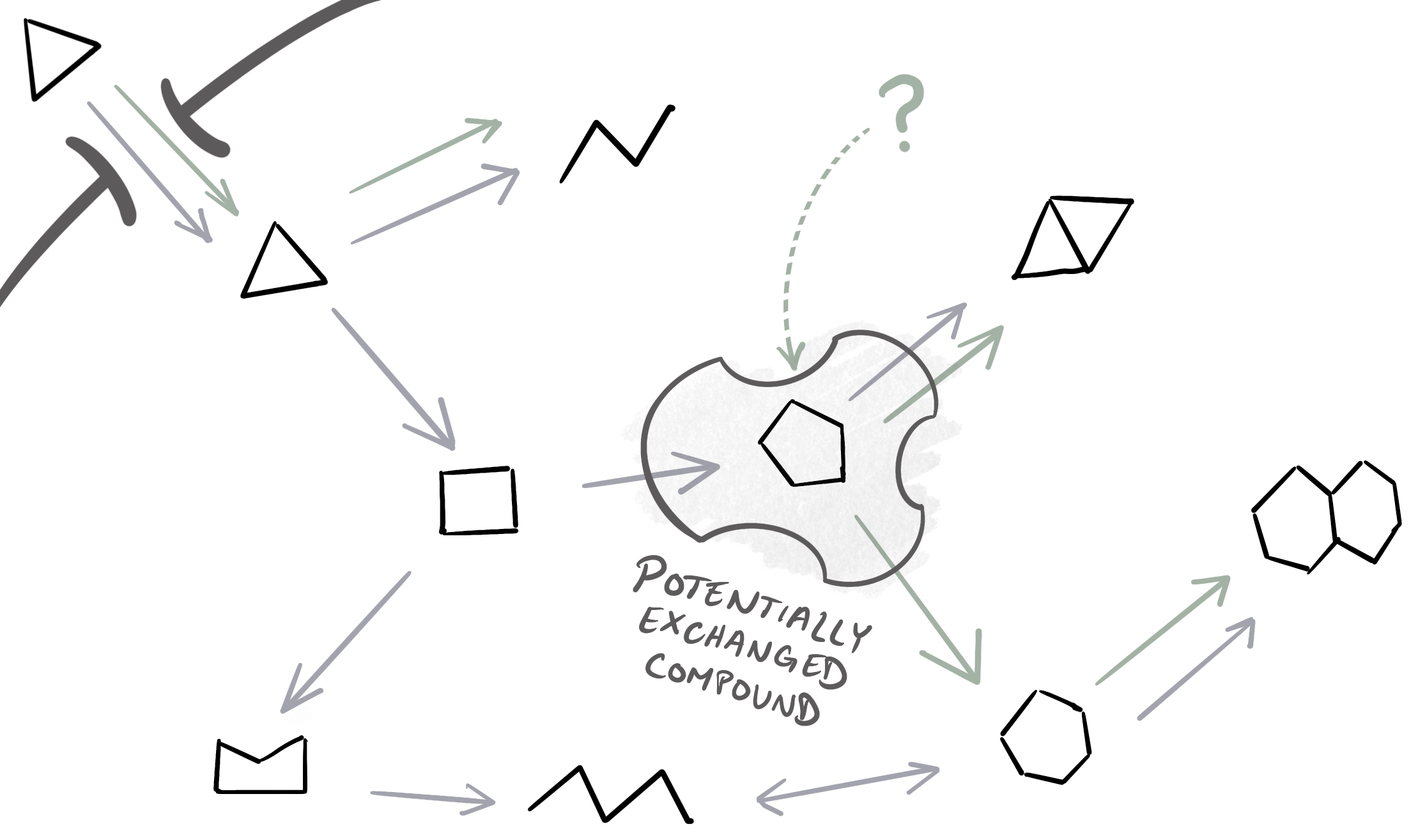
Essentially, the program examines the potential transfer point of the metabolite, meaning the point during the chemical transformations of the compound at which it could be transferred from one genome to another. However, there are two different strategies for examining this transfer point and its surrounding network of reactions, which result in predictions with different levels of accuracy and confidence. These two approaches are described in the next two sections (in order of their complexity).
The “Reaction Network Subset” approach
This approach implements almost exactly the ‘transfer point’ examination described in the previous section: for a given compound, we subset the merged reaction network to keep only the reactions immediately surrounding the compound (ie, production and/or consumption reactions). This allows us to designate the role of each genome using the following rules:
- if a genome’s reaction network includes a production reaction for the compound, then it is considered a ‘producer’
- if a genome’s reaction network includes a consumption reaction for the compound, then it is considered a ‘consumer’
- a genome can be both a ‘producer’ and a ‘consumer’, or it can be neither
Then we can check if only one genome can participate in those reactions (a ‘unique’ compound), if the production or consumption is one-sided (a ‘potentially-exchanged’ compound), or if we have one of those other situations from the table that we ignore (no prediction).
Advantages of this approach:
- it is relatively fast, because it is so simple and looks at only the potential ‘transfer point’ of a given compound.
- it covers more metabolites, because ModelSEED contains many compounds that are not described in KEGG Pathway Maps and the only way to predict exchanges of these metabolites is through this approach.
Caveats to this approach:
- it can lead to many, but potentially less-accurate predictions. Because this approach primarily relies on the chemical reaction and metabolite information stored in ModelSEED, which is (in our experience) more expansive yet less curated than KEGG, we have to be a bit careful with the information we use to make predictions, and this can lead to missing or spurious predictions. For example, we have observed that this strategy can fail to predict some known metabolic exchanges simply because there are no explicit ‘production’ (or ‘consumption’) reactions for the compound in ModelSEED – instead there might be a single reaction presented in one direction, but marked as a ‘reversible’ reaction in the database. We did try considering both possible directions for ‘reversible’ reactions in ModelSEED as part of this approach, yet doing so eliminated almost all predictions (especially for our test set of known exchanges) because the very expansive set of ‘reversible’ reactions made it look like each genome was both a producer and a consumer of everything. So in the current implementation of the Reaction Network Subset approach, we do not take reaction reversibility as described in ModelSEED into account (reaction reversibility in KEGG Pathway Maps, on the other hand, is considered as part of the Pathway Map Walk approach). Another example: ModelSEED can include many versions of the same or similar compound with separate compound ID numbers, which can prevent this approach from properly chaining together all reactions related to a given metabolite and can thus lead to mistakes (either predicting an exchange that is not real, or failing to identify an exchange). In theory, this issue can be circumvented by using
--custom-equivalent-compounds-fileto match all of the compound IDs that should be considered equivalent to each other. However, the scale of ModelSEED was too large for us to automatically identify matches for anything other than common amino acids (see the flag--use-equivalent-amino-acidsdescribed above). - there is no evidence associated with a prediction, and hence no way to filter the results. Since this approach only looks at the transfer point, and not upstream or downstream of that point within the reaction network, we don’t collect any contextual information that might be useful for interpreting the prediction – we don’t know if a given reaction is isolated within the network (potentially a spurious annotation) or part of a longer chain of reactions. In theory, we could be looking for this evidence by walking over the merged reaction network similar to the Pathway Map Walk strategy, but in practice, the number of possible paths through the network is too large for this to be computationally tractable (if we figure out a way to do it, we will certainly update the information here).
Bottom line: you have to be extra careful with interpreting any predictions labeled with the Reaction_Network_Subset prediction method in the output of this program.
Provided you run anvi-predict-metabolic-exchanges without the --pathway-walk-only flag, it will use the Reaction Network Subset approach. Normally, this strategy is the second stage of prediction. If you allow the Pathway Map Walk strategy to run, then that will run first to make a limited yet likely more accurate set of predictions for any compounds that participate in a KEGG Pathway Map. Afterwards, the Reaction Network Subset approach will process any reminaing compounds from the merged reaction network that do not participate in a KEGG Pathway Map.
If the Pathway Map Walk strategy is skipped (for instance, with the --no-pathway-walk), then the Reaction Network Subset approach will process all compounds from the merged reaction network.
The “Pathway Map Walk” approach
This prediction approach is more complex, utilizes additional information from the KEGG PATHWAY database, and adds evidence associated with each prediction that enables one to filter for more reasonable-looking results.
It starts by associating compounds from the merged reaction network with KEGG Pathway Maps, and for each of the resulting Pathway Maps, it finds all possible ‘walks’ through a Map for a given compound within the context of each genome’s reaction network. Briefly, a ‘walk’ starts from a compound and chains together the set of subsequent (or previous) reactions that are present in the genome’s network, until it finds a reaction that the genome cannot catalyze (due to the absence of required enzymes). A ‘walk’ can be done in either the ‘production’ direction (all reactions leading up to the production of the compound in the current organism) or the ‘consumption’ direction (all reactions leading away from the consumption of the compound in the current organism), and results in one or more reaction chains that describe the possible ways this metabolite can be generated or utilized by the organism. When there are branch points in the Pathway Map, all these branches are fully explored (potentially resulting in multiple reaction chains for either ‘production’ or ‘consumption’ of the compound). By default, we don’t allow any gaps in the chain (meaning that we stop exploring a branch once we reach a reaction that is not present in the reaction network), but you can change this using the parameter --maximum-gaps, in which case the walk will continue past these gaps until it reaches the maximum allowed number of missing reactions. The resulting reaction chains are specific to a given compound, Pathway Map, genome, and direction (‘production’ vs ‘consumption’).
To summarize, this Pathway Walk strategy gives us ‘production’ reaction chains and ‘consumption’ reaction chains for a compound within each organism. For example, if the compound in question is urea, and we are walking over Pathway Map map00230 (Purine Metabolism), we might end up with the following set of reaction chains:

In this example, the purple arrows represent the reactions found in the predicted ‘producer’ organism (who can generate urea from urate using a production chain of 5 reactions, and who cannot consume urea) while the green arrows represent the reactions found in the predicted ‘consumer’ organism. The consumer does not have a production chain because there are a lot of gaps in its network (in fact, we would not find a production chain in this genome unless we set --maximum-gaps to at least 3). However, it does have a consumption reaction chain of length 1 (the reaction converting urea to bicarbonate).
The Pathway Map walking functionality works directly with KGML files and was recently implemented by Dr. Sam Miller. It is a powerful subsystem of anvi’o that will hopefully be further described elsewhere in our documentation pages eventually. In the meantime, for the technically curious, you can check out the code in the kgml.py and kgmlnetworkops.py modules in the anvi’o Github repository.
Prediction of potential exchanges from the Pathway Map walk results is then fairly straightforward:
- if an organism has a ‘production’ chain for a compound from at least one Pathway Map, then it is considered a ‘producer’
- if an organism has a ‘consumption’ chain for a compound from at least one Pathway Map, then it is considered a ‘consumer’
- a genome can be both a ‘producer’ and a ‘consumer’, or it can be neither
And from there, we can decide which situation the compound falls into based on our prediction table, just like we do in the Reaction Network Subset approach. But unlike the Reaction Network Subset approach, we do not stop at prediction.
The final step of this approach is using the reaction chains returned by the Pathway Map walk to compute evidence that supports or opposes each prediction. We calculate the length of the longest production chain in the ‘primary producer’ of the compound, and compare this chain to any production chains in the ‘primary consumer’ to compute how much overlap there is (if any). We do the same thing on the other side of the transfer point, by computing the length of the longest consumption chain in the ‘primary consumer’ and evaluating the overlap from consumption chains in the ‘primary producer’. Overlap is described in both length form (number of overlapping reactions) and proportion form (relative to the length of the reaction chain). You can see examples of these values in the example reaction chain image above.
If a compound is found in multiple Pathway Maps, we aggregate the evidence from all of those maps and report the most informative Pathway Maps (one for production, one for consumption) in the main *-potentially-exchanged-compounds.txt output file. ‘Most informative’ here means the map with the longest reaction chain. And if there are multiple maps with the same maximum chain length, then we report the map with the smallest (real-number) proportion of overlap (‘real-number’ just means we try to avoid reporting None overlap values when possible). Detailed evidence from all Pathway Maps considered for a given compound is reported in the *-evidence.txt output file in case you need to take a closer look.
Advantages of this approach:
- higher confidence in the predictions. KEGG Pathway Maps are highly curated and tend to contain information about compounds and reactions that we can trust, leading us to make better decisions when predicting exchanges.
- evidence for filtering the results. For each potentially-exchanged compound, you can see whether the transfer point was surrounded by isolated reactions or longer chains of chemical transformations, whether the two organisms involved had lots of overlapping reactions related to the production (or consumption) of the compound, and which Pathway Maps this evidence comes from. All of these can help you decide whether a prediction looks reasonable or not.
Caveats to this approach:
- only a (relatively small) subset of known metabolites are described in KEGG Pathway Maps. We can’t make predictions for other compounds using this strategy.
- we cannot use all KEGG Pathway Maps for this strategy. A Pathway Map must have a ‘reaction’-type KGML file in order for us to do the walk, so we are forced to skip those that do not have such a file. (‘Reaction’-type KGML files have the prefix ‘RN’ for those that like to look at the database files themselves.) We explicitly exclude some Pathway Maps (for instance, map00470, which mostly makes connections between other, more specific maps), but for any map we do not explicitly exclude and which does not meet the requirement, you will likely see a warning on the terminal (these warnings are disabled in multi-mode, but you can use the
--debugflag to see if any Pathway Maps caused issues). - it is slow. Walking over Pathway Maps (especially big and/or complex Pathway Maps) is computationally intensive because it requires examining all possible branch points.
Bottom Line: you should probably focus on predictions from the prediction method Pathway_Map_Walk whenever possible
Provided you run anvi-predict-metabolic-exchanges without the --no-pathway-walk flag, it will use the Pathway Map Walk approach first to make predictions for any compound associated with at least one KEGG Pathway Map.
Which Pathway Maps do we use?
In order for the Pathway Map walk to work, we need the map to (1) include Kegg Orthologs (KOs) so that we can link up the reaction network information with the Pathway Map information, and (2) have a Reaction (‘RN’) type KGML file available in the KEGG data directory. This means we only process regular KEGG Pathway Maps categorized under ‘Metabolism’, whose map IDs start with the prefix ‘00’ – you can see a list of all Maps on the KEGG PATHWAY webpage if you are curious what falls under this category. And out of those maps, we have to skip any map that doesn’t have an ‘RN’-type KGML file. The program detects and skips any non-‘RN’ maps automatically and warns the user on the terminal about them.
If you want to exclude a subset of these KEGG Pathway Maps, you can do so with the --exclude-pathway-maps as described previously on this page.
Edit this file to update this information.
Additional Resources
Are you aware of resources that may help users better understand the utility of this program? Please feel free to edit this file on GitHub. If you are not sure how to do that, find the __resources__ tag in this file to see an example.