anvi-script-reformat-fasta
Table of Contents
Reformat FASTA file (remove contigs based on length, or based on a given list of deflines, and/or generate an output with simpler names).
🔙 To the main page of anvi’o programs and artifacts.
Authors



Requires
Provides
contigs-fasta ![]() contig-rename-report-txt
contig-rename-report-txt ![]()
Usage
A simpe program to perform a combination of simple operations on a FASTA file including,
- Renaming sequences so they have simplified deflines (more on this in the next section),
- Adding a prefix to sequence names in a FASTA file (useful when you wish to concatenate multiple FASTA files and want to make sure each sequence name is unique and tracable back to its original source),
- Removing sequences that are shorter or longer than specific thresholds, or only keeping those that match to a specific length,
- Removing sequences if they contain more than a number of gap characters or exceed the precentage of gap characters you permit (some simple quality checks prior to phylogenetic / phylogenomic analyses),
- Excluding sequences that match to a list of sequence IDs, or only keep those that match to a list of sequence IDs,
- Enforcing a sequence type and to replace any character with
Nfor nucleotide sequences that are not A, C, T, or G, or to replace any character withXfor amino acid sequences if the character does not match any of the single-letter amino acid characters (useuful to make sure the input file conforms the expectations of that input file type (i.e., all DNA sequences, or all AA sequences, etc)).
This program can work with compressed input FASTA files (i.e., the file name ends with a .gz extention) and will report a compressed output FASTA file (i.e., if the output file name ends with a .gz extension). It will just take awfully long time to run as it will have to decompress and recompress the file on the fly. But hey, you will have all the storage place you need to protect protected, right?
Stats-only mode
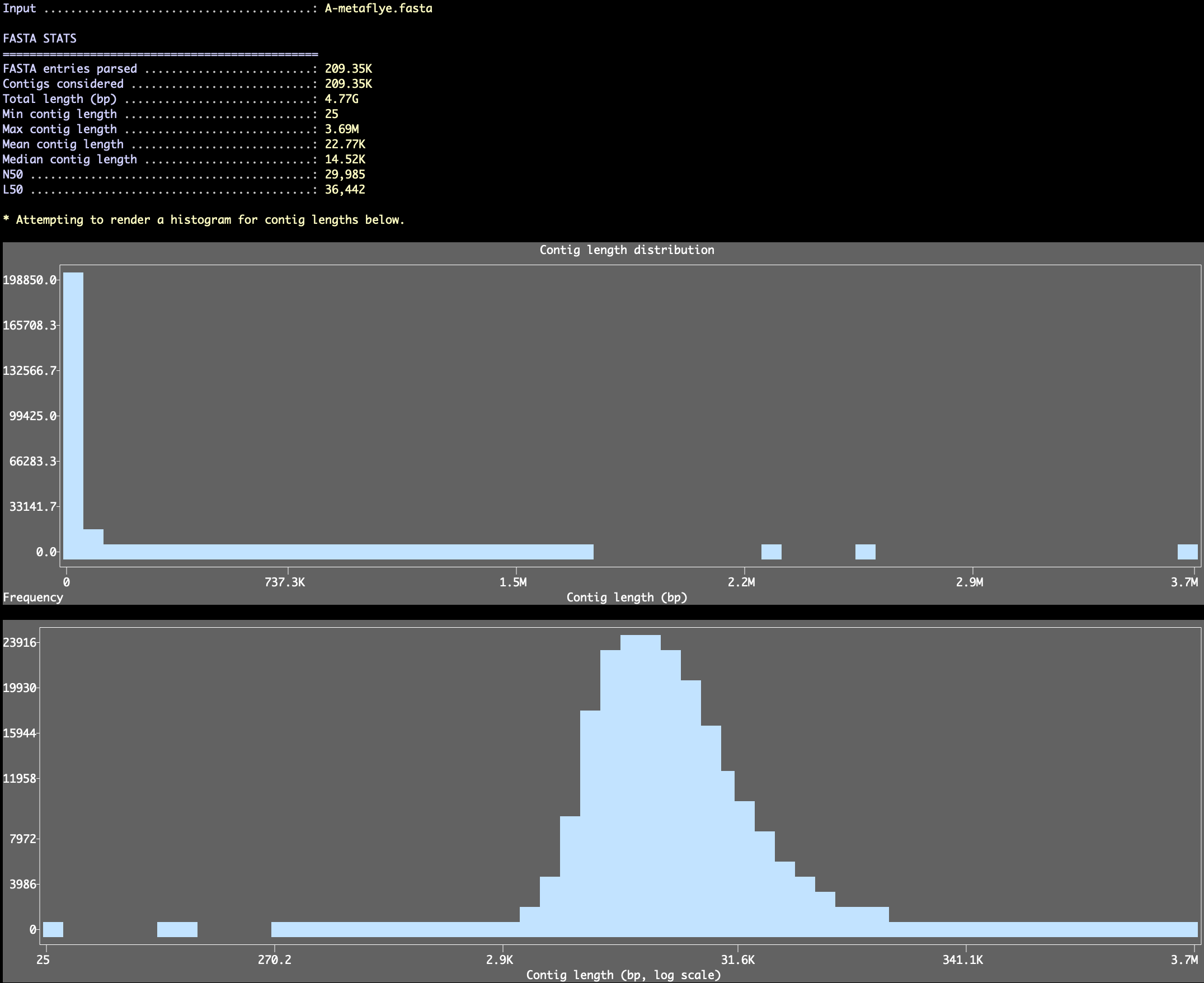
If you only want to take a quick look at FASTA-level summary statistics without writing a new file, you can ask the program to skip all reformatting steps and don’t change anything in the FASTA file with the --stats-only flag:
anvi-script-reformat-fasta fasta \ --stats-only
Which will report entry counts, length totals, min/max/mean/median lengths, and N50/L50, and render length histograms in the terminal (anvi’o will pick a bin count for these histograms, but you can also set a specific number using the --length-histogram-bins parameter and control the plot height with --length-histogram-height).
Renaming / simplifying sequence deflines
One of the most useful tasks this program performs is to simplify the deflines in your fasta file so they meet the conditions required of a contigs-fasta that is required by other anvi’o programs. You can simplify deflines in a fasta file the following way:
anvi-script-reformat-fasta fasta \ -o contigs-fasta \ --simplify-names \ --report-file contig-rename-report-txt
The --report-file flag is quite important to use here as it will generate a TAB-delimited file, contig-rename-report-txt, to keep track of which defline in the new file corresponds to which defline in the original file.
Removing the short reads
If your fasta file includes a lot of very short contigs, removing them may dramatically improve the performance of the generation and processing of your contigs-db. The example below runs the same command while also removing sequences that are shorter than 1,000 nts:
anvi-script-reformat-fasta fasta \ -o contigs-fasta \ -l 1000 \ --simplify-names \ --report-file contig-rename-report-txt
You can also discard unusually long sequences with the --max-len parameter if you want to enforce an upper bound on sequence size. You may ask yourself why would I discard my long contigs, but actually this can be quite useful when screening FASTA files for phylogenomic analyses, where you do not want to include sequences that are much longer than the expected siize of the model hits. To do it right, you can take a look at the histogram output, and determine what is your meaningful limits for length.
Example output
anvi-script-reformat-fasta contigs.fa \
--simplify-names \
--prefix YYY \
--min-len 1000 \
--seq-type NT \
--overwrite-input
Input ........................................: contigs.fa
Output .......................................: (anvi'o will overwrite your input file)
WHAT WAS THERE
===============================================
Total num contigs ............................: 4,189
Total num nucleotides ........................: 35,766,167
WHAT WAS ASKED
===============================================
Simplify deflines? ...........................: Yes
Add prefix to sequence names? ................: Yes, add 'YYY'
Minimum length of contigs to keep ............: 1,000
Maximum length of contigs to keep ............: No limit
Max % gaps allowed ...........................: 100.00%
Max num gaps allowed .........................: 1,000,000
Exclude specific sequences? ..................: No
Keep specific sequences? .....................: No
Enforce sequence type? .......................: Yes, enforce 'NT'
WHAT HAPPENED
===============================================
Contigs removed ..............................: 3,156 (75.34% of all)
Nucleotides removed ..........................: 6,121,239 (17.11% of all)
Nucleotides modified .........................: 161 (0.00045% of all)
Deflines simplified ..........................: True
* The contents of your input file have changed because you used the flag
`--overwrite-input`.
Please use the flag --overwrite-input with extreme caution.
Edit this file to update this information.
Additional Resources
Are you aware of resources that may help users better understand the utility of this program? Please feel free to edit this file on GitHub. If you are not sure how to do that, find the __resources__ tag in this file to see an example.