anvi-estimate-metabolism
Table of Contents
- Authors
- Can consume
- Can provide
- Usage
- What metabolism data can I use?
- Prerequisites to using this program
- Running metabolism estimation
- Estimation for a single genome
- Estimation for bins in a metagenome
- Estimation for a metagenome
- Estimation for a set of enzymes
- MULTI-MODE: Running metabolism estimation on multiple contigs databases
- Estimation for multiple single genomes
- Estimation for multiple bins
- Estimation for multiple metagenomes
- Adjustable Parameters
- Changing the module completion threshold
- Working with a non-default KEGG data directory
- Working with user-defined metabolism data
- Skipping KEGG data (ie, only working with user-defined metabolism data)
- Including KEGG Orthologs not in KOfam
- Output options
- Testing this program
- Help! I’m getting version errors!
- What to do if estimation is not working as expected for user-defined metabolic modules?
- Technical Details
- What data is used for estimation?
- Two estimation strategies - pathwise and stepwise
- How is pathwise completeness/copy number calculated?
- Part 1: Unrolling module definitions
- Part 2: Marking steps complete
- Part 3: Module completeness
- Part 4: Adjusting completeness
- Part 5: Path copy number
- Part 6: Module copy number
- Part 7: Adjusting copy number
- Pathwise Strategy Summary
- How is stepwise completeness/copy number calculated?
- Additional Resources
Reconstructs metabolic pathways and estimates pathway completeness for a given set of contigs.
🔙 To the main page of anvi’o programs and artifacts.
Authors

Can consume
contigs-db ![]() kegg-data
kegg-data ![]() kegg-functions
kegg-functions ![]() profile-db
profile-db ![]() collection
collection ![]() bin
bin ![]() external-genomes
external-genomes ![]() internal-genomes
internal-genomes ![]() metagenomes
metagenomes ![]() user-modules-data
user-modules-data ![]() enzymes-txt
enzymes-txt ![]()
Can provide
kegg-metabolism ![]() user-metabolism
user-metabolism ![]()
Usage
anvi-estimate-metabolism predicts the metabolic capabilities of organisms based on their genetic content. It relies upon kegg-functions and metabolism information from KEGG (kegg-data), which is stored in a modules-db. It can also use user-defined metabolic pathways, as described in user-modules-data.
The metabolic pathways that this program considers (by default) are those defined by KEGG Orthologs (KOs) in the KEGG MODULE resource. Each KO represents a gene function, and a KEGG module is a set of KOs that collectively carry out the steps in a metabolic pathway.
Alternatively or additionally, you can define your own set of metabolic modules and estimate their completeness with this program. Detailed instructions for doing this can be found by looking at the user-modules-data and anvi-setup-user-modules pages.
Given a properly annotated contigs-db, this program determines which enzymes are present and uses these functions to compute the completeness of each metabolic module. There are currently two strategies for estimating module completeness - pathwise and stepwise - which are discussed in the technical details section on this page. The output of this program is one or more tabular text files - see kegg-metabolism for the output description and examples.
For a practical tutorial on how to use this program, visit this link. A more abstract discussion of available parameters, as well as technical details about how the metabolism estimation is done, can be found below.
What metabolism data can I use?
You have three options when it comes to estimating metabolism.
- KEGG only (this is the default). In this case, estimation will be run on modules from the KEGG MODULES database, which you must set up on your computer using anvi-setup-kegg-data. If you have a default setup of KEGG, you need not provide any parameters to choose this option. However, if you have your KEGG data in a non-default location on your computer, you will have to use the
--kegg-data-dirparameter to point out its location. - KEGG + USER data. In this case, we estimate on KEGG modules as in (1), but also on user-defined metabolic modules that you set up with anvi-setup-user-modules and provide to this program with the
--user-modulesparameter. - USER data only. You can elect to skip estimation on KEGG modules and only run on your own data by providing both the
--user-modulesand--only-user-modulesparameters.
Prerequisites to using this program
Metabolism estimation relies on gene annotations from the functional annotation source ‘KOfam’, also referred to as kegg-functions. Therefore, for this to work, you need to have annotated your contigs-db with hits to the KEGG KOfam database by running anvi-run-kegg-kofams prior to using this program, unless you are using the --only-user-modules option to ONLY estimate on user-defined metabolic modules.
Both anvi-run-kegg-kofams and anvi-estimate-metabolism rely on the kegg-data provided by anvi-setup-kegg-data, so if you do not already have that data on your computer, anvi-setup-kegg-data needs to be run first. To summarize, these are the steps that need to be done before you can use anvi-estimate-metabolism:
- Run anvi-setup-kegg-data to get data from KEGG onto your computer. This step only needs to be done once.
- [If not using
--only-user-modules] Run anvi-run-kegg-kofams to annotate your contigs-db with kegg-functions. This program must be run on each contigs database that you want to estimate metabolism for.
If you want to estimate for your own metabolism data, then you have a couple of extra steps to go through:
- Define your own metabolic modules by following the formatting guidelines described here and here, and then run anvi-setup-user-modules to parse them into a modules-db,
- Annotate your contigs-db with the functional annotation sources that are required for your module definitions. This may require running a few different programs. For instance, if your modules are defined in terms of NCBI COGS (ie, the
COG20_FUNCTIONannotation source), you will need to run anvi-run-ncbi-cogs. If you are using a set of custom HMMs, you will need to run anvi-run-hmms on that set using the--add-to-functions-tableparameter. If you already have annotations from one or more of these sources, you could also import them into the contigs database using the program anvi-import-functions.
Running metabolism estimation
You can run metabolism estimation on any set of annotated sequences, but these sequences typically fall under one of the following categories:
- Single genomes, also referred to as external-genomes. These can be isolate genomes or metagenome-assembled genomes, for example. Each one is described in its own individual contigs-db.
- Bins, also referred to as internal-genomes. These often represent metagenome-assembled genomes, but generally can be any subset of sequences within a database. A single contigs-db can contain multiple bins.
- Assembled, unbinned metagenomes. There is no distinction between sequences that belong to different microbial populations in the contigs-db for an unbinned metagenome.
As you can see, anvi-estimate-metabolism takes one or more contigs database(s) as input, but the information that is taken from those databases depends on the context (ie, genome, metagenome, bin). In the case of internal genomes (or bins), is possible to have multiple inputs but only one input contigs db. So for clarity’s sake, we sometimes refer to the inputs as ‘samples’ in the descriptions below. If you are getting confused, just try to remember that a ‘sample’ can be a genome, a metagenome, or a bin.
If you don’t have any sequences, there is an additional input option for you:
- A list of enzymes, as described in an enzymes-txt file. For the purposes of metabolism estimation, the enzymes in this file will be interpreted as all coming from the same ‘genome’.
Different input contexts can require different parameters or additional inputs. The following sections describe what is necessary for each input type.
Estimation for a single genome
The most basic use-case for this program is when you have one contigs database describing a single genome. Since all of the sequences in this database belong to the same genome, all of the gene annotations will be used for metabolism estimation.
anvi-estimate-metabolism -c contigs-db
Estimation for bins in a metagenome
You can estimate metabolism for different subsets of the sequences in your contigs database if you first bin them and save them as a collection. For each bin, only the gene annotations from its subset of sequences will contribute to the module completeness scores.
You can estimate metabolism for every individual bin in a collection by providing the profile database that describes the collection as well as the collection name:
anvi-estimate-metabolism -c contigs-db -p profile-db -C collection
The metabolism estimation results for each bin will be printed to the same output file(s). The bin_name column in long-format output will distinguish between results from different bins.
If you only want estimation results for a single bin, you can instead provide a specific bin name from that collection using the -b parameter:
anvi-estimate-metabolism -c contigs-db -p profile-db -C collection -b bin
Or, to estimate on a subset of bins in the collection, you can provide a text file containing the specific list of bins that you are interested in:
anvi-estimate-metabolism -c contigs-db -p profile-db -C collection -B bin_ids.txt
Each line in the bin_ids.txt file should be a bin name from the collection (there is no header line). Here is an example file containing three bin names:
bin_1
bin_3
bin_5
Estimation for a metagenome
If you have an unbinned metagenome assembly, you can estimate metabolism for it using --metagenome-mode. In this case, since there is no way to determine which contigs belong to which microbial populations in the sample, estimation will be done on a per-contig basis; that is, for each contig, only the genes present on that contig will be used to determine pathway completeness within the contig.
anvi-estimate-metabolism -c contigs-db --metagenome-mode
In metagenome mode, this program will estimate metabolism for each contig in the metagenome separately. This will tend to underestimate module completeness because it is likely that many modules will be broken up across multiple contigs belonging to the same population. If you prefer to instead treat all enzyme annotations in the metagenome as belonging to one collective genome, you can do so by simply leaving out the --metagenome-mode flag (to effectively pretend that you are doing estimation for a single genome, although in your heart you will know that your contigs database really contains a metagenome). Please note that this will result in the opposite tendency to overestimate module completeness (as the enzymes will in reality be coming from multiple different populations), and there will be a lot of redundancy. We are working on improving our estimation algorithm for metagenome mode. In the meantime, if you are worried about the misleading results from either of these situations, we suggest binning your metagenomes first and running estimation for the bins as described below.
Estimation for a set of enzymes
Suppose you have a list of enzymes. This could be an entirely theoretical list, or they could come from some annotation data that you got outside of anvi’o - regardless of where you came up with this set, you can figure out what metabolic pathways these enzymes contribute to. All you have to do is format that list as an enzymes-txt file, and give that input file to this program, like so:
anvi-estimate-metabolism --enzymes-txt enzymes-txt
The program will pretend all of these enzymes are coming from one theoretical ‘genome’ (though the reality depends on how you defined or obtained the set), so the completion estimates for each metabolic pathway will consider all enzymes in the file. If you want to instead break up your set of enzymes across multiple ‘genomes’, then you will have to make multiple different input files and run this program on each one.
MULTI-MODE: Running metabolism estimation on multiple contigs databases
If you have a set of contigs databases of the same type (i.e., all of them are single genomes or all are binned metagenomes), you can analyze them all at once. What you need to do is put the relevant information for each contigs-db into a text file and pass that text file to anvi-estimate-metabolism. The program will then run estimation individually on each contigs database in the file. The estimation results for each database will be aggregated and printed to the same output file(s).
One advantage that multi-mode unlocks is the ability to generate matrix-formatted output, which is convenient for clustering or visualizing the metabolic potential of multiple samples. See the Output options section below for more details.
Estimation for multiple single genomes
Multiple single genomes (also known as external-genomes) can be analyzed with the same command by providing an external genomes file to anvi-estimate-metabolism. To see the required format for the external genomes file, see external-genomes.
anvi-estimate-metabolism -e external-genomes.txt
Estimation for multiple bins
If you have multiple bins (also known as internal-genomes), they can be analyzed with the same command by providing an internal genomes file to anvi-estimate-metabolism. The bins in this file can be from the same collection, from different collections, or even from different metagenomes. To see the required format for the internal genomes file, see internal-genomes.
anvi-estimate-metabolism -i internal-genomes.txt
Estimation for multiple metagenomes
Multiple metagenomes can be analyzed with the same command by providing a metagenomes input file. Metagenome mode will be used to analyze each contigs database in the file. To see the required format for the metagenomes file, see metagenomes.
anvi-estimate-metabolism -M metagenomes.txt
Adjustable Parameters
There are many ways to alter the behavior of this program to fit your needs. You can find some commonly adjusted parameters below. For a full list of parameters, check the program help (-h) output.
Changing the module completion threshold
As explained in the technical details section below, module completeness is computed as the percentage of steps in the metabolic pathway that are ‘present’ based on the annotated enzymes in the contigs database. If this completeness is larger than a certain percentage, then the entire module is considered to be ‘complete’ in the sample and the corresponding row in the long-format modules mode output file will have ‘True’ under the module_is_complete column. By default, the module completion threshold is 0.75, or 75%.
Changing this parameter usually doesn’t have any effect other than changing the proportions of ‘True’ and ‘False’ values in the module_is_complete column of long-format modules mode output (or the proportion of 1s and 0s in the module presence-absence matrix for --matrix-format output). It does not alter completeness scores. It also does not affect which modules are printed to the output file, unless you use the --only-complete flag (described in a later section). Therefore, the purpose of changing this threshold is usually so that you can filter the output later somehow (i.e., by searching for ‘True’ values in the long-format output).
The one exception is when --add-copy-number is used. We use the module completeness threshold to determine pathwise copy number of a module, which is based on the number of complete copies of paths through a module. So if you change this threshold, you can expect to see some differences in pathwise copy number values (which are found in certain long-format and matrix output files).
In this example, we change the threshold to 50 percent.
anvi-estimate-metabolism -c contigs-db --module-completion-threshold 0.5
Working with a non-default KEGG data directory
If you have previously annotated your contigs databases using a non-default KEGG data directory with --kegg-data-dir (see anvi-run-kegg-kofams), or you have moved the KEGG data directory that you wish to use to a non-default location, then you will need to specify where to find the KEGG data so that this program can use the right one. In that case, this is how you do it:
anvi-estimate-metabolism -c contigs-db --kegg-data-dir /path/to/directory/KEGG
Working with user-defined metabolism data
If you have defined your own set of metabolic modules and generated a modules-db for them using anvi-setup-user-modules, you can estimate the completeness of these pathways (in addition to the KEGG modules) by providing the path to the directory containing this data:
anvi-estimate-metabolism -c contigs-db --user-modules /path/to/USER/directory
The --user-modules parameter can be used in conjunction with the --kegg-data-dir parameter to control which KEGG data is being used at the same time.
Skipping KEGG data (ie, only working with user-defined metabolism data)
If you wish to only estimate for your own metabolic modules, you can skip estimating for KEGG modules by providing the --only-user-modules flag. The nice thing about doing this is that you can skip running anvi-run-kegg-kofams on your databases (which will save you lots of time and computational resources).
anvi-estimate-metabolism -c contigs-db --user-modules /path/to/USER/directory --only-user-modules
Including KEGG Orthologs not in KOfam
Sometimes, your input data may have annotations for KOs that are not in the KOfam profiles that we use for annotation. This can happen if you are using enzymes-txt, or if you have imported external annotations with the source name KOfam. By default, we don’t consider these annotations, and you will probably see an error message. However, (as suggested in that message) you can explicitly include these non-KOfam annotations into the analysis by providing the flag --include-kos-not-in-kofam, like so:
anvi-estimate-metabolism --enzymes-txt enzymes-txt --include-kos-not-in-kofam
Output options
This program has two types of output files: long-format (tab-delimited) output files and matrices. The long-format output is the default. If you are using multi-mode to work with multiple samples, you can request matrix output by using the flag --matrix-format.
You can find more details on the output formats by looking at kegg-metabolism. Below, you will find examples of how to use output-related parameters.
Long-Format Output
Long-format output has several preset “modes” as well as a “custom” mode in which the user can define the contents of the output file. Multiple modes can be used at once, and each requested “mode” will result in a separate output file. The default output mode is “modules” mode.
Viewing available output modes
To see what long-format output modes are currently available, use the --list-available-modes flag:
anvi-estimate-metabolism -c contigs-db --list-available-modes
The program will print a list like the one below and then exit.
AVAILABLE OUTPUT MODES
===============================================
modules ......................................: Information on metabolic modules
modules_custom ...............................: A custom tab-delimited output file where you choose the included modules data using --custom-output-headers
module_paths .................................: Information on each possible path (complete set of enzymes) in a module
module_steps .................................: Information on each top-level step in a module
hits .........................................: Information on all enzyme annotations in the contigs DB, regardless of module membership
Please note that you must provide your input file(s) for this to work.
Using a non-default output mode
You can specify one or more long-format output modes using the --output-modes parameter. The mode names must exactly match to one of the available modes from the --list-available-modes output.
anvi-estimate-metabolism -c contigs-db --output-modes hits
Using multiple output modes
If you want more than one output mode, you can provide multiple comma-separated mode names to the --output-modes parameter. There should be no spaces between the mode names.
anvi-estimate-metabolism -c contigs-db --output-modes hits,modules
When multiple output modes are requested, a different output file is produced for each mode. All output files will have the same prefix, and the file suffixes specify the output mode. For example, modules mode output has the suffix _modules.txt while hits mode has the suffix _hits.txt.
Viewing available output headers for ‘custom’ mode
The modules_custom output mode allows you to specify which information to include (as columns) in your long-format output. It is essentially a customizable version of modules mode output. To use this mode, you must specify which columns to include by listing the column names after the --custom-output-headers flag.
To find out what column headers are available, use the --list-available-output-headers parameter:
anvi-estimate-metabolism -c contigs-db --list-available-output-headers
The program will print a list like the one below and then exit.
AVAILABLE OUTPUT HEADERS
===============================================
module .......................................: Module number [modules output mode]
stepwise_module_is_complete ..................: Whether a module is considered complete or not based on its STEPWISE percent completeness and the completeness threshold [modules output mode]
stepwise_module_completeness .................: Percent completeness of a module, computed as the number of complete steps divided by the number of total steps (where 'steps' are determined by splitting the module definition
on the space character) [modules output mode]
pathwise_module_is_complete ..................: Whether a module is considered complete or not based on its PATHWISE percent completeness and the completeness threshold [modules output mode]
pathwise_module_completeness .................: Percent completeness of a module, computed as maximum completeness of all possible combinations of enzymes ('paths') in the definition [modules output mode]
enzymes_unique_to_module .....................: A list of enzymes that only belong to this module (ie, they are not members of multiple modules) [modules output mode]
unique_enzymes_hit_counts ....................: How many times each unique enzyme appears in the sample (order of counts corresponds to list in `enzymes_unique_to_module` field) [modules output mode]
proportion_unique_enzymes_present ............: Proportion of enzymes unique to this one module that are present in the sample [modules output mode]
unique_enzymes_context_string ................: Describes the unique enzymes contributing to the `proportion_unique_enzymes_present` field [modules output mode]
module_name ..................................: Name/description of a module [modules output mode]
[....]
As you can see, this flag is also useful when you want to quickly look up the description of each column of data in your output files.
For each header, the output mode(s) that it is applicable to are listed after the description. The headers you can choose from for modules_custom output end in either [modules output mode] or [all output modes].
Just as with --list-available-modes, you must provide your input file(s) for this to work. In fact, some headers will change depending on which input types you provide. You will see additional possible headers if you use the --add-copy-number or --add-coverage flags (though this only works for single sample inputs, not for Multi Mode - if you wish to get custom output for Multi Mode, it is best to construct your custom header list by looking at the possible headers for your given parameter set for a SINGLE sample from your input file).
Using custom output mode
Here is an example of defining the modules output to contain columns with the module number, the module name, and the completeness score. The corresponding headers for these columns are provided as a comma-separated list (no spaces) to the --custom-output-headers flag.
anvi-estimate-metabolism -c contigs-db --output-modes modules_custom --custom-output-headers module,module_name,module_completeness
Including modules with 0% completeness in long-format output
By default, modules with completeness scores of 0 are not printed to the output files to save on space (both pathwise completeness and stepwise completeness must be 0 to exclude modules from the output). But you can explicitly include them by adding the --include-zeros flag.
anvi-estimate-metabolism -c contigs-db --include-zeros
Including module copy number in long-format output
You can ask this program to count the number of copies of each module in your input samples by providing the --add-copy-number flag:
anvi-estimate-metabolism -c contigs-db --output-modes modules,module_paths,module_steps --add-copy-number
Just like module completeness, copy number can be calculated using two different strategies. You can find information about the calculations in the technical details section below, and information about what copy number output looks like in kegg-metabolism.
This flag also works for matrix output.
Including coverage and detection in long-format output
If you have a profile database associated with your contigs database and you would like to include coverage and detection data in the metabolism estimation output files, you can use the --add-coverage flag. You will need to provide the profile database as well, of course. :)
anvi-estimate-metabolism -c contigs-db -p profile-db --output-modes modules,hits --add-coverage
This option also works for the --enzymes-txt input option, provided that you include both a coverage column and a detection column in the enzymes-txt input file.
anvi-estimate-metabolism --enzymes-txt enzymes-txt --add-coverage
For hits mode output files, in which each row describes one enzyme annotation for a gene in the contigs database, the output will contain two additional columns per sample in the profile database. One column will contain the mean coverage of that particular gene call by reads from that sample and the other will contain the detection of that gene in the sample.
For modules mode output files, in which each row describes a metabolic module, the output will contain four additional columns per sample in the profile database. One column will contain comma-separated mean coverage values for each gene call in the module, in the same order as the corresponding gene calls in the gene_caller_ids_in_module column. Another column will contain the average of these gene coverage values, which represents the average coverage of the entire module. Likewise, the third and fourth columns will contain comma-separated detection values for each gene call and the average detection, respectively.
Note that you can customize which coverage/detection columns are in the output files if you use custom modules mode. Use the following command to find out which coverage/detection headers are available:
anvi-estimate-metabolism -c contigs-db -p profile-db --add-coverage --list-available-output-headers
Matrix Output
Matrix format is only available when working with multiple contigs databases. Several output matrices will be generated, each of which describes one statistic such as module completion score, module presence/absence, or enzyme annotation (hit) counts. As with long-format output, each output file will have the same prefix and the file suffixes will indicate which type of data is present in the file.
In each matrix, the rows will describe modules, top-level steps, or enzymes. The columns will describe your input samples (i.e. genomes, metagenomes, bins), and each cell will be the corresponding statistic. You can see examples of this output format by viewing kegg-metabolism.
Obtaining matrix-formatted output
Getting these matrices is as easy as providing the --matrix-format flag.
anvi-estimate-metabolism -i internal-genomes.txt --matrix-format
Including metadata in the matrix output
By default, the matrix output is a matrix ready for use in other computational applications, like visualizing as a heatmap or performing clustering. That means it has a header line and an index in the right-most column, but all other cells are numerical. However, you may want to instead have a matrix that is annotated with more information, like the names and categories of each module or the functional annotations of each enzyme. To include this additional information in the matrix output (as columns that occur before the sample columns), use the --include-metadata flag.
anvi-estimate-metabolism -i internal-genomes.txt --matrix-format --include-metadata
Note that this flag only works for matrix output because, well, the long-format output inherently includes metadata.
Including rows of all zeros in the matrix output
The --include-zeros flag works for matrix output, too. By default, modules that have 0 completeness (or enzymes that have 0 hits) in every input sample will be left out of the matrix files. Using --include-zeros results in the inclusion of these items (that is, the inclusion of rows of all zeros).
anvi-estimate-metabolism -i internal-genomes.txt --matrix-format --include-zeros
Getting module-specific enzyme matrices
The standard enzyme hit matrix includes all enzymes that were annotated at least once in your input databases (or all enzymes that we know about, if --include-zeros is used). But sometimes you might want to see a matrix with only the enzymes from a particular metabolic pathway. To do this, pass a comma-separated (no spaces) list of module numbers to the --module-specific-matrices flag, and then your matrix output will include enzyme hit matrices for each module in the list.
For example,
anvi-estimate-metabolism -e input_txt_files/external_genomes.txt \ --matrix-format \ --module-specific-matrices M00001,M00009 \ -O external_genomes
will produce the output files external_genomes-M00001_enzyme_hits-MATRIX.txt and external_genomes-M00009_enzyme_hits-MATRIX.txt (in addition to the typical output matrices). Each additional output matrix will include one row for each enzyme in the module, in the order it appears in the module definition. It will also include comment lines for each major step (or set of steps) in the module definition, to help with interpreting the output.
Check out this (partial) example for module M00001:
enzyme isolate E_faecalis_6240 test_2
# (K00844,K12407,K00845,K25026,K00886,K08074,K00918)
K00844 0 0 0
K12407 0 0 0
K00845 0 0 0
K25026 0 1 0
K00886 1 0 1
K08074 0 0 0
K00918 0 0 0
# (K01810,K06859,K13810,K15916)
K01810 1 1 1
K06859 0 0 0
K13810 0 0 0
K15916 0 0 0
# (K00850,K16370,K21071,K00918)
K00850 0 1 0
K16370 0 0 0
K21071 0 0 0
K00918 0 0 0
[....]
If you don’t want those comment lines in there, you can combine this with the --no-comments flag to get a clean matrix. This might be useful if you want to do some downstream processing of the matrices.
anvi-estimate-metabolism -e input_txt_files/external_genomes.txt \ --matrix-format \ --module-specific-matrices M00001,M00009 \ --no-comments \ -O external_genomes
In this case, the above file would look like this:
enzyme isolate E_faecalis_6240 test_2
K00844 0 0 0
K12407 0 0 0
K00845 0 0 0
K25026 0 1 0
K00886 1 0 1
K08074 0 0 0
K00918 0 0 0
K01810 1 1 1
K06859 0 0 0
K13810 0 0 0
K15916 0 0 0
K00850 0 1 0
K16370 0 0 0
K21071 0 0 0
K00918 0 0 0
[....]
Including copy number in matrix output
The --add-copy-number flag, which was discussed above for including module copy number values in long-format output, also works for matrix output:
anvi-estimate-metabolism -i internal-genomes.txt --matrix-format --add-copy-number
When you use this flag, you will get matrices describing copy number statistics in addition to the typical set of matrix output files.
Other output options
Regardless of which output type you are working with, there are a few generic options for controlling how the output files look like.
Changing the output file prefix
anvi-estimate-metabolism can produce a variety of output files. All will be prefixed with the same string, which by default is kegg-metabolism. If you want to change this prefix, use the -O flag.
anvi-estimate-metabolism -c contigs-db -O my-cool-prefix
Including only complete modules in the output
Remember that module completion threshold? Well, you can use that to control which modules make it into your output files. If you provide the --only-complete flag, then any module-related output files will only include modules that have a completeness score (either pathwise or stepwise) at or above the module completion threshold. (This doesn’t affect enzyme-related outputs, for obvious reasons.)
Here is an example of using this flag with long-format output (which is the default, as described above, but we are asking for it explicitly here just to be clear):
anvi-estimate-metabolism -c contigs-db --output-modes modules --only-complete
And here is an example of using this flag with matrix output. In this case, we are working with multiple input samples, and the behavior of this flag is slightly different: a module will be included in the matrix if it is at or above the module completion threshold in at least one sample, for either pathwise or stepwise completeness. That means you may see numbers lower than the threshold in the completeness matrices.
anvi-estimate-metabolism -i internal-genomes.txt --matrix-format --only-complete
Testing this program
You can see if this program is working on your computer by running the following suite of tests, which will check several common use-cases:
anvi-self-test --suite metabolism
Help! I’m getting version errors!
If you have gotten an error that looks something like this:
Config Error: The contigs DB that you are working with has been annotated with a different version of the MODULES.db than you are working with now.
This means that the modules-db used by anvi-run-kegg-kofams has different contents (different KOs and/or different modules) than the one you are currently using to estimate metabolism, which would lead to mismatches if metabolism estimation were to continue. There are a few ways this can happen:
- You upgraded to a new anvi’o version and downloaded the default kegg-data associated with that release, but are working with a contigs-db that was annotated with a previous anvi’o version (and therefore a different instance of kegg-data).
- Without changing anvi’o versions, you annotated your contigs-db with default kegg-data, and subsequently replaced that data with a different instance by running anvi-setup-kegg-data again with the
--resetflag (and likely also with the--kegg-archive,--kegg-snapshot, or--download-from-keggoptions, all of which get you a non-default version of KEGG data). Then you tried to run anvi-estimate-metabolism with the new data. - You have multiple instances of kegg-data on your computer in different locations, and you used different ones for anvi-run-kegg-kofams and anvi-estimate-metabolism.
- Your collaborator gave you some databases that they annotated with a different version of kegg-data than you have on your computer.
There are two main solutions for most of these situations, which differ according to which set of annotations you would prefer to use.
First option: you want to update your contigs-db to have new annotations that match to the current modules-db. In this case, you have to rerun anvi-run-kegg-kofams on the contigs-db. Make sure you provide the same --kegg-data-dir value (if any) that you put in the anvi-estimate-metabolism command that gave you this error.
Second option: you want to continue working with the existing set of annotations in the contigs-db. This means you need to change which modules-db you are using for anvi-estimate-metabolism. The error message should tell you the hash of the modules-db used for annotation. You can use that hash to identify the matching database so that you can either re-download that database, or (if you already have it) find it on your computer.
If you have multiple instances of kegg-data on your computer, you can run anvi-db-info on the modules-db in each of those directories until you find the one with the hash you are looking for. Then provide the path to that directory using the kegg-data-dir parameter of anvi-estimate-metabolism.
If you’ve recently upgraded your anvi’o version (i.e., situation 1 from above) and you kept your previous installation of anvi’o, the database you want should still be available as part of that environment. You can find its location by activating the environment and running the following code in your terminal: export ANVIO_MODULES_DB=python -c “import anvio; import os; print(os.path.join(os.path.dirname(anvio.file), ‘data/misc/KEGG/MODULES.db’))”``. Use echo $ANVIO_MODULES_DB to print the path in your terminal, and anvi-db-info $ANVIO_MODULES_DB to verify that its hash matches to the one in your contigs database.
If you don’t have any matching instances of kegg-data on your computer, you will need to download it. First, check if the version you want is one of the KEGG snapshots that anvi’o provides by looking at the KEGG-SNAPSHOTS.yaml file in the anvi’o codebase. For instance, you can get the location of that file and print it to your terminal by running the following:
export ANVIO_KEGG_SNAPSHOTS=python -c "import anvio; import os; print(os.path.join(os.path.dirname(anvio.__file__), 'data/misc/KEGG-SNAPSHOTS.yaml'))"
cat $ANVIO_KEGG_SNAPSHOTS`.
Take a look through the different versions. If you see one with a hash matching to the one used to annotate your contigs-db, then you can download that version by following the directions for setting up a KEGG snapshot. Provide the snapshot version name to the --kegg-snapshot parameter of anvi-setup-kegg-data.
I can’t find KEGG data with a matching hash!
If you don’t have a matching metabolism database on your computer, and none of the snapshots in the KEGG-SNAPSHOTS.yaml file have the hash that you need, your contigs-db was probably annotated with KO and module data downloaded directly from KEGG. If you have obtained the contigs-db from a collaborator (i.e., situation 4 from above), ask them to also share their kegg-data with you, following these steps. Otherwise, anvi’o cannot really help you get this data back, and you may have to resort to option 1 described above.
If none of these solutions help you to get rid of the version incompatibility error, please feel free to reach out to the anvi’o developers for help.
What to do if estimation is not working as expected for user-defined metabolic modules?
If you are estimating completeness of user-defined modules and find that the results are not as expected, you should double check your module files to make sure the pathway is defined properly. Are the enzyme accession numbers in the DEFINITION correct? Do you have the proper ANNOTATION_SOURCE for each enzyme, and are these lines spelled properly and matching to the annotation sources in your contigs database(s)? If you are using custom HMM profiles, did you remember to use the --add-to-functions-table parameter?
If these things are correct but you are still not finding an annotation for one or more enzymes that you know should be in your sequence data, consider why those annotations might not be there - perhaps the e-values are too low for the annotations to be kept in the database? Keep in mind that you can always try to add enzyme annotations (with the proper sources) to your database using anvi-import-functions before running anvi-estimate-metabolism again.
Technical Details
What data is used for estimation?
Regardless of which input type is provided to this program, the basic requirements for metabolism estimation are 1) a set of metabolic pathway definitions, and 2) a ‘pool’ of gene annotations.
Module Definitions
One set of metabolic pathway definitions that can be used by this program is the KEGG MODULE resource. You can also define your own set of metabolic modules, but the definition format and estimation strategy will be the same. So for brevity’s sake, the following discussion will cover the KEGG data case.
The program anvi-setup-kegg-data acquires the definitions of these modules using the KEGG API and puts them into the modules-db. The definitions are strings of KEGG Ortholog (KO) identifiers, representing the functions necessary to carry out each step of the metabolic pathway. Let’s use module M00018, Threonine Biosynthesis, as an example. Here is the module definition, in picture form:
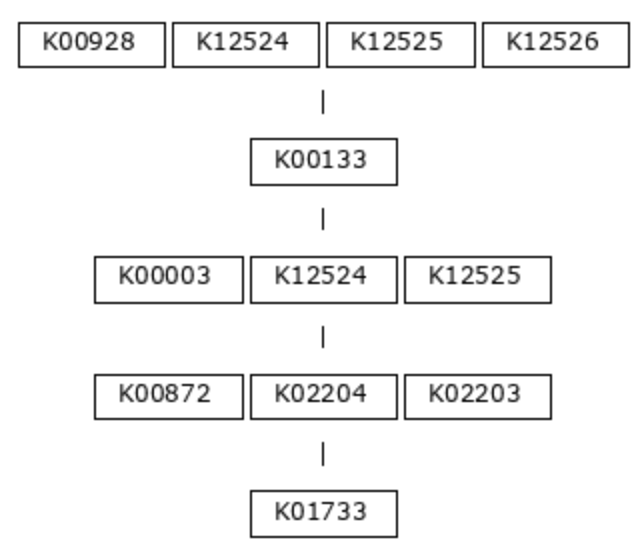
This biosynthesis pathway has five major steps, or chemical reactions (we call these major steps ‘top-level steps’, which will be important later). The first reaction in the pathway requires an aspartate kinase enzyme (also known as a homoserine dehydrogenase), and there are four possible orthologs known to encode this function: K00928, K12524, K12525, or K12526. Only one of these genes is required to be able to carry out this step. In contrast, the second reaction can be fulfilled by only one known KO, the aspartate-semialdehyde dehydrogenase K00133.
The definition string for module M00018 is this:
(K00928,K12524,K12525,K12526) K00133 (K00003,K12524,K12525) (K00872,K02204,K02203) K01733
Hopefully the correspondence between the picture and text is clear - spaces separate distinct steps in the pathway, while commas separate alternatives.
That was a simple example, so let’s look at a more complicated one: M00011, the second carbon oxidation phase of the citrate cycle.
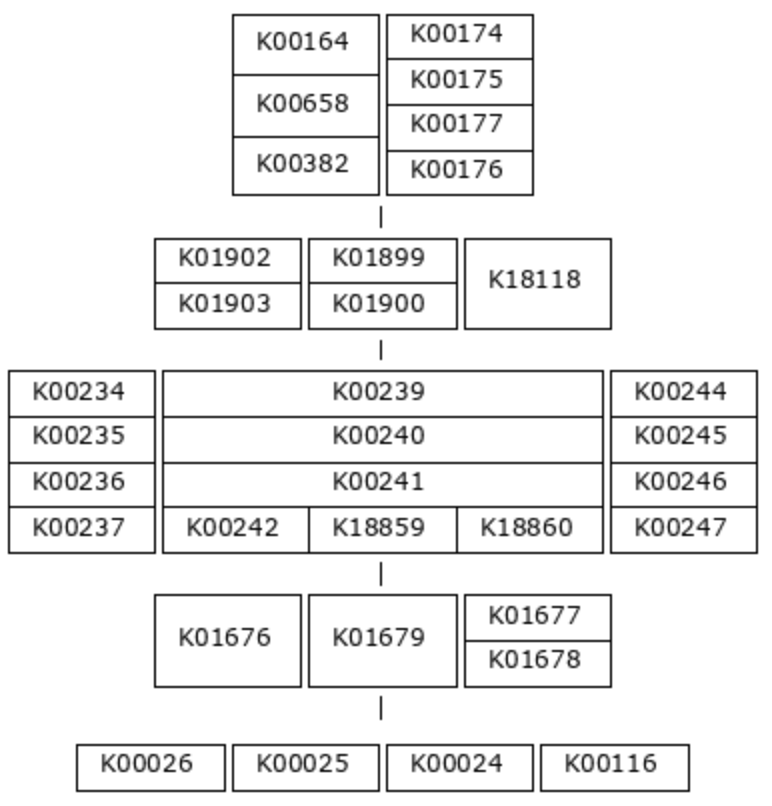
This pathway also has five steps, but this time, most of the reactions require an enzyme complex. Each KO within a multi-KO box is a component of an enzyme. For example, one option for the first reaction is 2-oxoglutarate dehydrogenase, a 3-component enzyme made up of K00164, K00658, and K00382.
This is the definition string for module M00011:
(K00164+K00658+K00382,K00174+K00175-K00177-K00176) (K01902+K01903,K01899+K01900,K18118) (K00234+K00235+K00236+K00237,K00239+K00240+K00241-(K00242,K18859,K18860),K00244+K00245+K00246-K00247) (K01676,K01679,K01677+K01678) (K00026,K00025,K00024,K00116)
And here is a detail that is difficult to tell from the pictorial definition - not all enzyme components are equally important. You can see in the definition string that KO components of an enzyme complex are connected with either ‘+’ signs or ‘-‘ signs. The ‘+’ sign indicates that the following KO is an essential component of the enzyme, while the ‘-‘ sign indicates that it is non-essential. For the purposes of module completeness estimation, we only consider a reaction to be fulfilled if all the essential component KOs are present in the annotation pool (and we don’t care about the ‘non-essential’ components). So, for example, we would consider the first step in this pathway complete if just K00174 and K00175 were present. The presence/absence of either K00177 or K00176 would not affect the module completeness score at all.
Module definitions can be even more complex than this. Both of these examples had exactly five top-level steps, no matter which set of KOs you use to fulfill each reaction. However, in some modules, there can be alternative sets with different numbers of steps. In addition, some modules (such as M00611, the module representing photosynthesis), are made up of other modules, in which case they are only complete if their component modules are complete.
Hopefully this information will help you understand our estimation strategies in the next section.
KOfam (enzyme) annotations
For metabolism estimation to work properly, gene identifiers in the pool of annotations must match to the gene identifiers used in the pathway definitions. For KEGG MODULEs, we rely on annotations from the KEGG KOfam database, which is a set of HMM profiles for KEGG Orthologs (KOs). The program anvi-run-kegg-kofams can annotate your contigs-db with hits to the KEGG KOfam database. It adds these annotations under the source name ‘KOfam’.
Which of the annotations are considered for metabolism estimation depends on the input context. If you are working with isolate genomes (ie, not metagenome mode or bins), then all of the annotations under source ‘KOfam’ will be used. If you are working with bins in metagenomes, then for each bin, only the ‘KOfam’ annotations that are present in that bin will be in the annotation pool. Finally, for metagenome mode, since estimation is done for each contig separately, only the annotations present in each contig will be considered at a time.
User-defined metabolic modules must specify the annotation source(s) needed to find their component enzymes in your data. Adding these annotation sources to your contigs databases may require running a variety of programs. However, anvi-estimate-metabolism loads these gene annotations and uses them in the same way as it does ‘KOfam’ annotations for KEGG data.
Two estimation strategies - pathwise and stepwise
We currently have two ways of estimating the completeness of a module, which differ in how we decompose the module DEFINITION string into smaller parts.
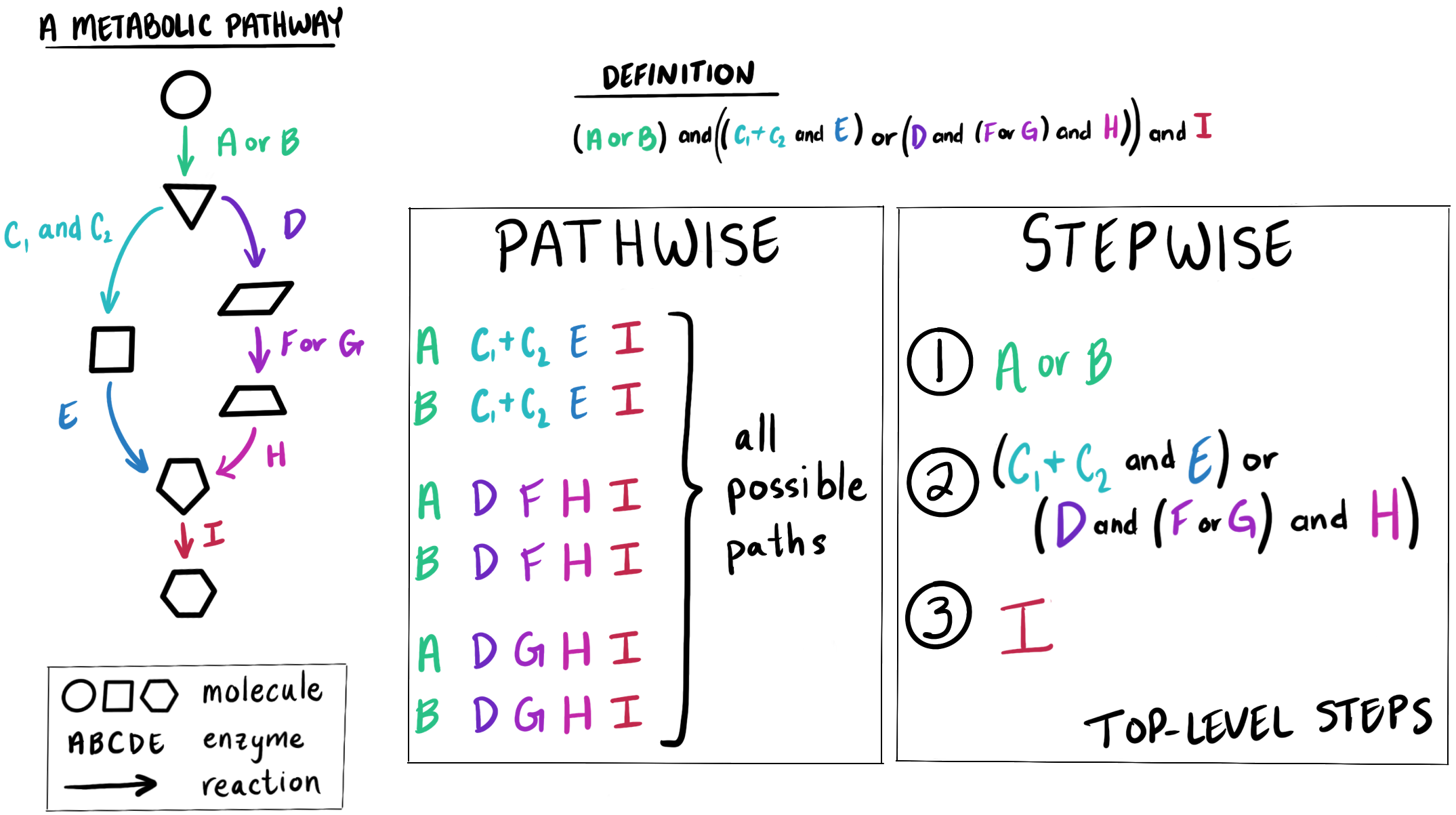
For the ‘pathwise’ strategy, we consider all possible ‘paths’ through the module - each alternative set of enzymes that could be used together to catalyze every reaction in the metabolic pathway. After calculating the percent completeness in all possible paths, we take the maximum completeness to be the pathwise completeness score of the module as a whole. This is the most granular way of estimating module completeness because we consider all the possible alternatives.
For the ‘stepwise’ strategy, we break down the module DEFINITION into its major, or ‘top-level’, steps. Each “top-level” step usually represents either one metabolic reaction or a branch point in the pathway, and is defined by one or more enzymes that either work together or serve as alternatives to each other to catalyze this reaction or set of reactions. We use the available enzyme annotations to determine whether each step can be catalyzed or not - just a binary value representing whether the step is present or not. Then we compute the stepwise module completeness as the percent of present top-level steps. This is the least granular way of estimating module completeness because we do not distinguish between enzyme alternatives - these are all considered as one step which is either entirely present or entirely absent.
The pathwise and stepwise strategies also apply to copy number calculations, in which enzyme annotations are allocated to create different copies of a path, step, or module. Path copy number is computed as the number of complete copies of a path through a module, and a module’s pathwise copy number is then calculated as the maximum copy number of any of its paths that have the highest completeness score. Step copy number is the number of complete copies of a top-level step, and a module’s stepwise copy number is the minimum copy number of all of its top–level steps.
Confused? Yeah, this is complicated stuff! But hopefully the illustrative examples in the next few sections will clear it up.
How is pathwise completeness/copy number calculated?
For demonstration purposes, let’s talk through the estimation of pathwise completeness and copy number for one module, in one ‘sample’ (ie a genome, bin, or contig in a metagenome). Just keep in mind that the steps described below are followed for each module in each sample.
Part 1: Unrolling module definitions
As you saw above in the module examples, there can be multiple alternative KOs for a given step in a pathway. This means that there can be more than one way to have a ‘complete’ metabolic module. Therefore, to estimate completeness, we first have to identify all possible ‘paths’ through the module definition, where a ‘path’ is a set of KOs that could make the module complete (if they were all present in the annotation pool).
anvi-estimate-metabolism uses a recursive algorithm to “unroll” the module definition string into a list of all possible paths. First, the definition string is split into its top-level steps (which are separated by spaces). Each step is either an atomic step, a protein complex (KO components separated by ‘+’ or ‘-‘), or a compound step (multiple alternatives, separated by commas). Compound steps and protein complexes are recursively broken down until we have only atomic steps. An atomic step can be a single KO, a module number, a nonessential KO starting with ‘-‘, or -- (a string indicating that there is a reaction for which we do not have a KO). We use the atomic steps to build a list of alternative paths through the module definition. Protein complexes are split into their respective components using this strategy to find all possible alternative complexes, and then these complexes (with all their component KOs) are used to build the alternative paths.
Let’s see this in action, using the Threonine Biosynthesis module from above as an example. We first split the definition on spaces to get all top-level steps. Here we show each top-level step on its own line:
(K00928,K12524,K12525,K12526)
K00133
(K00003,K12524,K12525)
(K00872,K02204,K02203)
K01733
The first step is made up of 4 alternative KOs. We split on the commas to get these, and thus we have the starting KO for 4 possible alternative paths:
K00928 K12524 K12525 K12526
| | | |
The second step, K00133, is already an atomic step, so we can simply extend each of the paths with this KO:
K00928 K12524 K12525 K12526
| | | |
K00133 K00133 K00133 K00133
The third step is another compound step, but this time we can get 3 atomic steps out of it. That means that our 4 possible paths so far each gets 3 alternatives, bringing our total alternative path count up to 12:
K00928 K12524 K12525 K12526
| | | |
K00133 K00133 K00133 K00133
/ | \ / | \ / | \ / | \
K00003 K12524 K12525 K00003 K12524 K12525 K00003 K12524 K12525 K00003 K12524 K12525
Okay, hopefully you get the picture by now. The end result is a list of lists, like this:
[[K00928,K00133,K00003,K00872,K01733],
[K00928,K00133,K00003,K02204,K01733],
......
[K12526,K00133,K12525,K02203,K01733]]
in which every inner list is one of the alternative paths through the module definition - one of the possible ways to have a complete module.
By the way, here is one alternative path from the module M00011, just so you know what these look like with protein complexes:
[K00164+K00658+K00382,K01902+K01903,K00234+K00235+K00236+K00237,K01676,K00026]
Part 2: Marking steps complete
Once we have our list of alternative paths through the module, the next task is to compute the completeness of each path. Each alternative path is a list of atomic steps or protein complexes. We loop over every step in the path and use the annotation pool of KOs to decide whether the step is complete (1) or not (0). We have the following cases to handle:
-
A single KO - this is easy. If we have an annotation for this KO in our pool of ‘KOfam’ annotations, then the step is complete (1).
-
A protein complex - remember that these are multiple KOs connected with ‘+’ (if they are essential components) or ‘-‘ (if they are non-essential). Well, for these steps, we compute a fractional completeness based on the number of essential components that are present in the annotation pool. We basically ignore the non-essential KOs. For example, the complex ‘K00174+K00175-K00177-K00176’ would be considered 50% complete (a score of 0.5) if only ‘K00174’ were present in the annotation pool.
-
Non-essential KOs - some KOs are marked as non-essential even when they are not part of a protein complex. They look like this: ‘-K12420’, with a minus sign in front of the KO identifier (that particular example comes from module M00778). These steps are ignored for the purposes of computing module completeness.
-
Steps without associated KOs - some reactions do not have a KO identifier, but instead there is the string
--serving as a placeholder in the module definition. Since we can’t annotate the genes required for these steps, we have no idea if they are complete or not, so we always consider them incomplete (0). Modules that have steps like this can therefore never have 100% completeness - it is sad, but what can we do? We warn the user about these instances so that they can check manually for any missing steps. -
Modules - finally, some modules are defined by other modules. We can’t determine if these steps are complete until we’ve estimated completeness for every module, so we ignore these for now.
To get the completeness score for a given path through the module, we first add up the completeness of each essential step in the path and then we divide that sum by the number of essential steps.
Part 3: Module completeness
By this time, we have a completeness score (a fraction between 0 and 1) for every possible path through the module. To get the completeness score for the module overall, we simply take the maximum of all these completeness scores.
Why take the maximum? We are assuming here that if the metabolic pathway is actually being used in a cell (which we can’t know for sure without doing some transcriptomics and possibly metabolomics), the most complete set of enzymes in that pathway is the most likely to be used. This is certainly a questionable assumption, but we need to make some choices like this in order to summarize the data, so we do it. It gets tricker to interpret this number when there is more than one path through the module that has the maximum completeness score - which one is being used? We cannot know just from (meta)genomics data, so it would take additional data types or knowledge of the biological system to figure this out.
We can then check this number against the module completeness threshold (which is 0.75 by default). If the module completeness score is greater than or equal to the threshold, we mark the module as ‘complete’. This boolean value is meant only as a way to easily filter through the modules output, and you shouldn’t put too much stock in it because it covers up a lot of nuances, as you can tell from the details above :).
Note that some modules, especially those with a lot of possible paths, can have more than one path which has the maximum completeness score (that is, the score that determines the completeness of the module). This will be important later for calculating pathwise copy number, so we keep track of all of the paths with this maximum completeness score.
Part 4: Adjusting completeness
But don’t forget that there are some modules defined by other modules. These are usually what KEGG calls ‘Signature Modules’, which are collections of enzymes that collectively encode some phenotype, rather than a typical pathway of chemical reactions. For these modules, we have to go back and adjust the completeness score after we know the completeness of its component modules. To do this, we basically re-do the previous two tasks to recompute the number of complete steps in each path and the overall completeness of the module. This time, when we reach a ‘Module’ atomic step (case 5), we take that module’s fractional completeness score to be the completeness of the step.
As an example, consider module M00618, the Acetogen signature module. Its definition is
M00377 M00579
Suppose module M00377 had a completeness score of 0.7 and module M00579 had a score of 0.4, based on the prior estimations. Then the completeness score of the [M00377,M00579] path would be (0.7+0.4)/2 = 0.55. Since this is the only possible path through the module, M00618 is 55% complete.
Part 5: Path copy number
In reality, this part is actually done at the same time as Part 2, but it is easier to understand if we think of it separately. We have all our possible paths through the module from Part 1, and we need to figure out the copy number of each path. To do this, we look at the number of annotations of each atomic step in the path. Here is how we handle each atomic step:
-
A single KO - the copy number of this atomic step is equal to the number of annotations (hits) of this enzyme. If the atomic step is K00133 and you have 5 genes annotated with K00133, then you have 5 copies of that step. If you have 0 annotations, then the step copy number is 0. This is the simplest case.
-
A protein complex - these are a bit tricky. Once again, we ignore the non-essential components and only consider essential ones. But we use the number of annotations to figure out how many complete copies of the protein complex exist in the input sample. For example, suppose we have 2 annotations for K00174 and 3 annotations for K00175. These are the only two essential components in the complex ‘K00174+K00175-K00177-K00176’. Since we have at least two annotations for both of these enzymes, we have two copies of the complex. What about the third annotation for K00175? Well, it can’t do much all by itself. This hypothetical third copy of the complex is only 50% complete (1 out of 2 essential components), which is less than the default module completeness threshold of 75%. So we ignore it, and say that we have 2 copies of the enzyme complex. However, this calculation can change if you were to adjust the module completeness threshold. If you set the threshold to be 50% or lower, then 50% is enough to consider the third copy of the complex complete (in which case, we would say that we have 3 copies of the enzyme complex).
-
Non-essential KOs - just like we ignore these steps when computing completeness, we also ignore them when computing copy number.
-
Steps without associated KOs (the
--case) - Just like we always consider these atomic steps to be incomplete, we also always give them a copy number of 0. -
Modules - as you might guess, the copy number of these atomic steps are obtained later, after we’ve computed copy number for every other module. There is an adjustment step for copy number just like there is one for completeness (Part 4 above).
To get the copy number for a given path through the module, we determine the number of complete copies of the path. This is the same as the way we handle copy number of protein complexes, as described above. And it also depends on the module completeness threshold. Suppose a path has 4 essential atomic steps (call them A,B,C, and D) with the following copy numbers: 4,3,1, and 2. Using the default completeness threshold of 0.75, we need at least 3 out of 4 atomic steps to be present in order for a copy of the path to be considered complete. There is one copy that has all 4 steps, one copy that has 3/4, one copy that has 2/4 and one that has 1/4. This is perhaps easier to see in graph form, with atomic steps on the x-axis and atomic step copy number on the y-axis:
X
X X
X X X <-- (3/4 enzymes present)
X X X X <-- (4/4 enzymes present)
A B C D
Each copy of the path is a horizontal row of X’s in the simple graphic above. There are 2 copies of the path with at least 3/4 atomic steps in the list (marked with arrows), which means that the path copy number is 2.
Part 6: Module copy number
Once we have the completeness scores and copy numbers of all possible paths through the module, we can compute the copy number of the module itself. Remember from Part 3 that we saved the paths which have the highest completeness score? We take the maximum copy number of those paths of highest completeness.
So if the module does not have any complete paths, then its copy number is 0. If it has one complete path, then its copy number is the copy number of that path. If there are multiple paths with highest completeness score, then its copy number is the maximum of the copy numbers of those paths - for example, let’s say we have two paths, both of which are 90% complete. One of those paths has a copy number of 1 and the other has a copy number of 3. The module copy number would be 3 in this case.
We’re making assumptions here again, just like we were when computing module completeness. Any of those paths (or none of them) could be the one that is used in the cell, and we don’t know which one. But the idea here is that if a sample has the most copies of path X, there is probably a good reason that is has that many copies because microbial cells like to streamline their genomes whenever possible.
One last note - if a module does not have any paths of highest completeness, we cannot compute the copy number. In this case, the copy number of the module will be reported as ‘NA’ in the output file(s).
Part 7: Adjusting copy number
This part is analogous to Part 4, in that we go back later to adjust the copy number of modules defined by other modules. We set the copy number of a module atomic step to be the previously-computed copy number of that module (if any). The tricky bit here is that some modules can have a copy number of ‘NA’. When this is the case for one of our atomic steps, we make the adjusted module copy number ‘NA’ as well.
Pathwise Strategy Summary
In short: pathwise module completeness in a given sample is calculated as the maximum fraction of essential KOs (enzymes) that are annotated in the sample, where the maximum is taken over all possible sets of KOs (enzymes) from the module definition. Likewise, pathwise module copy number is calculated as the maximum copy number of any path with the module’s completeness score.
These values get harder to interpret when we are considering metagenomes rather than the genomes of individual organisms. There could be lots of different paths through a module used by different populations in a metagenome, but the module completeness/copy number values would summarize only the most common path(s). For situations like this, it is a good idea to take advantage of the ‘module_paths’ output mode to look at these scores for all individual paths through each module.
How is stepwise completeness/copy number calculated?
Now we’ll walk through an example of estimating stepwise completeness and copy number in one sample, again keeping in mind that the following steps are repeated for each module in each sample.
Part 1: Top-level steps
For stepwise completeness and copy number, we do our calculations at the level of top-level steps. These are the major steps in a metabolic pathway, each of which usually represents a single chemical reaction. A top-level step describes the enzyme(s) that can be used to catalyze that reaction. We can get the top-level steps of a module by splitting its DEFINITION string by its spaces (not including any spaces within parentheses).
Let’s use module M00018 as an example again. Earlier we described how M00018 is made up of five major steps, each one of which represents a single reaction in this metabolic pathway.
We take the M00018 DEFINITION string:
(K00928,K12524,K12525,K12526) K00133 (K00003,K12524,K12525) (K00872,K02204,K02203) K01733
and split it by spaces to get the top-level steps:
(K00928,K12524,K12525,K12526)
K00133
(K00003,K12524,K12525)
(K00872,K02204,K02203)
K01733
This is far more straightforward than unrolling the module into all possible paths. For the stepwise metrics, we will focus only on these major steps, and what’s more, we will ignore a lot of the nuance that comes from alternative enzymes within a top-level step.
Part 2: Step completeness
Unlike pathwise completeness, where we consider all possible alternatives and compute a fractional completeness for each path, a top-level step can only be entirely complete (1) or entirely incomplete (0). In other words, step completeness is binary. We don’t care how the step is complete. It doesn’t matter which of the enzymes in a step are used to make it complete.
To compute this binary completeness for each top-level step, we convert the step into a Boolean expression by following this set of rules:
- enzyme accessions (ie, KOs) are replaced with ‘True’ if the enzyme is annotated in the sample, and otherwise are replaced with ‘False’.
--steps do not have associated enzyme profiles, so we cannot say whether these steps are complete. These are always ‘False’.- commas represent alternative enzymes, meaning you can use either one or the other. We convert commas into OR relationships.
- spaces represent sequential enzymes, meaning that you need both (one after the other). We convert spaces into AND relationships.
- plus signs (‘+’) represent essential enzyme components, meaning that you need both (at the same time). We convert plus signs into AND relationships.
- minus signs (‘-‘) represent nonessential enzyme components, meaning that you don’t need them. We ignore these.
- parentheses are kept where they are to maintain proper order of operations.
After this conversion is done, we can simply evaluate the Boolean expression to determine whether or not the step is complete.
Let’s use the step (K00928,K12524,K12525,K12526) from M00018 as an example. Suppose that both K12524 and K12526 are annotated in the sample. Then this step would be converted into the following Boolean expression:
(False OR True OR False OR True)
which evaluates to True, meaning that this step is complete (1).
Here’s a more complicated example from module M00849:
((K00869 (K17942,(K25517+K09128 K25518+K03186))),(K18689 K18690 K22813))
Suppose the following enzymes are annotated in the sample: K00869, K25517, K09128, K25518, K18689, and K22813. Then this step would become the following Boolean expression:
((True AND (False OR (True AND True AND True AND False))) OR (True AND False AND True))
Since this is a bit more complicated, we must evaluate it by following order of operations:
=> ((True AND (False OR (False))) OR (True AND False AND True))
=> ((True AND (False)) OR (True AND False AND True))
=> (False) OR (True AND False AND True))
=> (False) OR (False)
=> False
Since it ultimately evaluates to False, this step is incomplete (0).
Note: if a top-level step includes entire modules in its definition, we skip evaluating its completeness for now.
Part 3: Module completeness
Once we’ve evaluated the binary completeness of each top-level step in a module, we calculate the stepwise completeness of the module by simply taking the percentage of complete top-level steps. So if, for instance, our five top-level steps in M00018 were evaluated like this:
(K00928,K12524,K12525,K12526) => complete (1)
K00133 => complete (1)
(K00003,K12524,K12525) => not complete (0)
(K00872,K02204,K02203) => complete (1)
K01733 => complete (1)
Then the overall stepwise completeness of M00018 would be 4/5, or 80%. If we were using the default module completeness threshold of 0.75, then this module would be considered ‘complete’ overall based on its stepwise score.
Note: if any of the module’s top-level steps are defined by other modules, we skip computing its completeness for now because we don’t know the completeness of these steps yet. These will be adjusted in the next section.
Part 4: Adjusting completeness
Just like in pathwise completeness, we need to deal with the case when a module is defined by another module. In Part 3, we skipped any modules that have top-level steps defined by other modules. Now, we go back and re-compute the completeness of any of these steps using the Boolean expression from Part 2 (ie, by replacing module accessions with ‘True’ if their stepwise completeness is above the module completeness threshold, or ‘False’ otherwise). Then we repeat Part 3 on the affected modules to calculate their overall stepwise completeness.
Part 5: Step copy number
Now we need to calculate the copy number of each top-level step. We can do this by converting the step into an arithmetic expression this time, by following a new set of rules:
- enzyme accessions (ie, KOs) are replaced with the number of annotations this accession has in the given sample.
--steps are unknown, so we replace these with a count of ‘0’.- commas represent alternative enzymes, meaning you can use either one or the other. We convert commas into addition operations.
- spaces represent sequential enzymes, meaning that you need both (one after the other). We convert spaces into min() operations.
- plus signs (‘+’) represent essential enzyme components, meaning that you need both (at the same time). We convert plus signs into min() operations.
- minus signs (‘-‘) represent nonessential enzyme components, meaning that you don’t need them. We ignore these.
- parentheses are kept where they are to maintain proper order of operations.
By doing it this way, we take into account all possible ways to complete the step without caring about which of the enzymes are contributing.
Let’s go through our examples again, this time with enzyme hit counts. For (K00928,K12524,K12525,K12526), suppose that K12524 was annotated once and K12526 was annotated twice in the sample. Then this step becomes the following arithmetic expression:
(0 + 1 + 0 + 2)
=> 3
This evaluates to a step copy number of 3. K12524 and K12526 can catalyze the same reaction, so their hit counts are combined when we compute the copy number.
Now our complex example, ((K00869 (K17942,(K25517+K09128 K25518+K03186))),(K18689 K18690 K22813)). Suppose that K00869 was annotated twice, K25517/K09128/K25518 were each annotated once, K03186 was annotated twice, and K18689 was annotated once. The conversion is a little bit more complicated here because we now need min() operations, so let’s go through it step-by-step. We do this by following the order of operations - so the innermost set of parentheses are converted first.
First, for the sequential enzyme complexes in (K25517+K09128 K25518+K03186), we require all four enzyme components to be present. So the minimum number of annotations of any of these four components determines the copy number of this part of the step. In other words, this combo is only as strong as its weakest link: min(K25517,K09128,K25518,K03186) becomes min(1,1,1,2) when we replace the enzyme accessions with their hit counts. We might have 2 copies of component K03186, but because we are limited by the copy numbers of the other three components, we only have 1 copy of this sub-step overall.
The comma in (K17942,(K25517+K09128 K25518+K03186) indicates that we can use either K17942 OR the sequential complexes (K25517+K09128 K25518+K03186). We’ve already converted the latter set of enzymes, so all that is left is to add the hit count of enzyme K17942 (which happens to be annotated 0 times): (0 + min(1,1,1,2)).
To finish up the left side of the step definition, we have an AND relationship between K00869 and the other enzymes in (K00869 (K17942,(K25517+K09128 K25518+K03186))). Since we need both, we take the minimum of K00869’s annotation count and of the expression we already converted: min(2,(0 + min(1,1,1,2))).
Moving on to the second half of the step definition, the sequential enzymes in (K18689 K18690 K22813) become min(1,0,0).
Finally, we put everything all together, using addition since there is a comma (OR relationship) between the two halves of the definition:
(min(2,(0 + min(1,1,1,2))) + min(1,0,0))
Here is the evaluation of the resulting arithmetic expression:
=> (min(2,(0 + 1)) + min(1,0,0))
=> (min(2,1) + min(1,0,0))
=> (1 + min(1,0,0))
=> (1 + 0)
=> 1
Ultimately, this step has a copy number of 1. This happened because there was at least one copy of every enzyme in the first half of the step definition (though you wouldn’t be able to figure this out just by looking at the step copy number).
Fun fact: this conversion from definition string to arithmetic expression is quite complex for a computer to do, and in the code for this program, it is implemented as a recursive function.
Part 6: Module copy number
Every top-level step in the module is connected by an AND relationship - you need all of the steps in order to have the module complete. For this reason, we compute the module stepwise copy number by taking the minimum copy number of all top-level steps. So if we had the following copy numbers for each top-level step in M00018:
(K00928,K12524,K12525,K12526) => 1 copy
K00133 => 2 copies
(K00003,K12524,K12525) => 0 copies
(K00872,K02204,K02203) => 1 copy
K01733 => 1 copy
Then the overall stepwise copy number of M00018 would be 0 (because the third step has 0 copies).
To make stepwise copy number easier to interpret, the output files of this program will include the individual step copy numbers in addition to the overall module copy number.
Part 7: Adjusting copy number
Once again, we must go back and adjust the copy number for any modules that are defined by other modules. For any top-level step whose definition includes modules, we take the arithmetic expression from Part 5 and replace those module accessions with the pre-computed stepwise copy number of the module. After evaluating the expression to get the copy number of these top-level steps, we can repeat Part 6 to get the module copy number.
Stepwise Strategy Summary
In short: stepwise module completeness in a given sample is calculated as the percentage of complete top-level steps. Likewise, stepwise module copy number is calculated as the minimum copy number of all top-level steps in the module definition.
To help interpret these stepwise metrics for modules, it is a good idea to look at the ‘module_steps’ output mode to see the scores for all individual top-level steps in a module.
Edit this file to update this information.
Additional Resources
Are you aware of resources that may help users better understand the utility of this program? Please feel free to edit this file on GitHub. If you are not sure how to do that, find the __resources__ tag in this file to see an example.